Nonetheless, these safeguards can be breached, especially as organisms age, and the consequences are often fatal. Prion and amyloid formation are associated with some of the most devastating neurodegenerative diseases confronting humankind, including Alzheimer’s disease, Parkinson's disease, variant Creutzfeldt-Jakob disease, and Huntington's disease (Cushman et al., 2010; Jackrel & Shorter, 2011). Yet, surprisingly, it is becoming increasingly clear that prions and amyloids are not always a problem. In fact, several have been harnessed during evolution for adaptive purposes and feature in some of the most revolutionary new concepts in biology and evolution, including protein-based genetic elements, long-term memory formation, melanosome biogenesis, evolutionary capacitance and the revelation of cryptic genetic variation (Shorter & Lindquist, 2005; Watt et al., 2009; Shorter, 2010). We employ biochemistry and genetics to understand the enigmatic mechanistic interfaces that exist between protein disaggregases, molecular chaperones, small molecules and amyloid/prion fibers or other misfolded species, and how these interfaces can be manipulated to divert pathogenic and promote beneficial phenotypic trajectories. Specifically, we are pursuing the following goals:
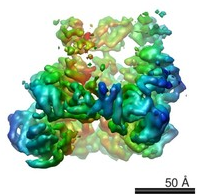
Hsp104
(Surface view colored by cylindrical radius)
click to videos of Hsp104
1) Defining the structural and mechanistic basis for Hsp104 function. One major focus concerns Hsp104, a protein disaggregase of the AAA+ superfamily from yeast, which disaggregates denatured proteins and returns them to normal function (Shorter, 2008; Vashist et al. 2010; DeSantis & Shorter, 2012a). Hsp104 is also essential for the formation and inheritance of several yeast prions; protein-based genetic elements comprised of amyloid fibers that self-perpetuate alterations in protein form and function. Hsp104 can both construct and deconstruct self-replicating amyloid forms of Sup35, which comprise the yeast prion [PSI+], and Ure2, which comprise the yeast prion [URE3] (Shorter & Lindquist, 2004; Shorter & Lindquist, 2006). We aim to elucidate the hexameric structure of Hsp104 (Wendler et al., 2007; Wendler et al., 2009; Sweeny et al., 2011; DeSantis et al., 2014; Sweeny et al., 2015). We also strive to understand the mechanistic basis of how Hsp104 structure enables these disaggregation activities and other prion-regulatory functions (DeSantis et al., 2012; DeSantis and Shorter, 2012b; Torrente et al., 2014; Sweeny et al., 2015).
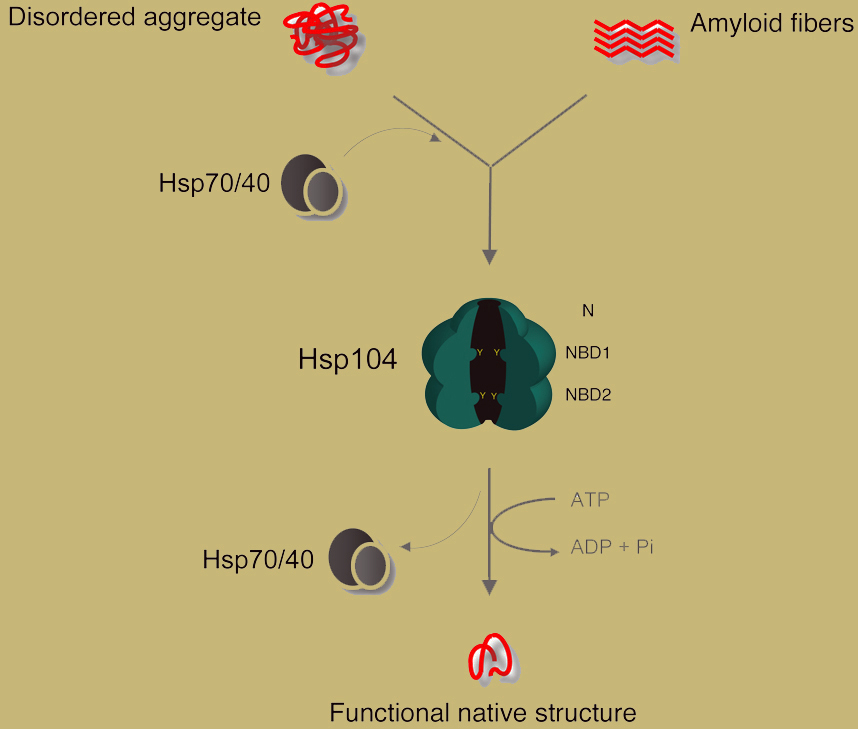
Hsp104 is often assisted by a supporting cast of molecular chaperones to rescue aggregated polypeptides (Sweeny & Shorter, 2008). Most notably, Hsp110, Hsp70, Hsp40 and small heat shock proteins synergize with Hsp104 to promote the reactivation of protein aggregates (Shorter, 2011; Duennwald et al., 2012). We wish to understand how these molecular chaperones achieve these synergistic activities (Doyle et al., 2007; Shorter & Lindquist, 2008; DeSantis & Shorter, 2012a; Duennwald et al., 2012; DeSantis et al., 2014). Finally, we seek to define the natural substrates comprising the Hsp104 folding reservoir and determine the impact of specific Hsp104 clients on yeast biology. [top]
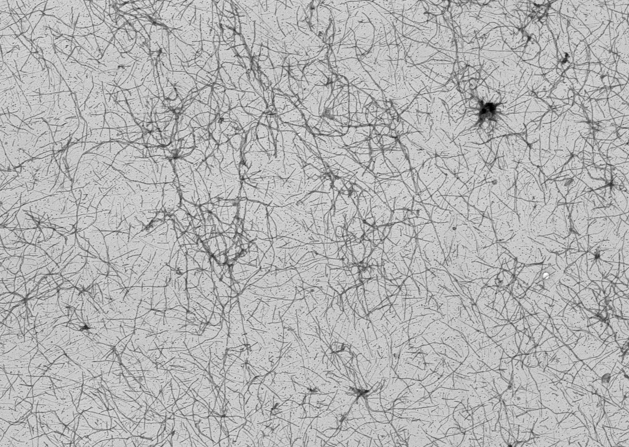
Sup35 fibers + Hsp104, t = 0 (Shorter & Lindquist, 2006)

Sup35 fibers + Hsp104, t = 20 minutes (Shorter & Lindquist, 2006)
2) Applying Hsp104 to disease-associated protein misfolding and aggregation. Inexplicably, Hsp104 has no known homologue or orthologue in metazoa. This deficiency is vexing, for it would seem that a protein that reverses protein aggregation and restores protein function, would be critical in our fight against several diseases caused by aberrant protein aggregation. Hence, we engineer, evolve and apply Hsp104 to metazoan systems to antagonize and reverse the proteotoxic aggregation pathways that are intimately connected with Parkinson’s, Alzheimer’s and Huntington’s disease as well as amyotrophic lateral sclerosis and HIV infection (Lo Bianco et al., 2008; Shorter, 2008; Vashist et al. 2010; Castellano and Shorter, 2012; DeSantis et al., 2012; Cushman-Nick et al., 2013; Jackrel et al., 2014a; Jackrel & Shorter, 2014a; Jackrel & Shorter, 2014b; Jackrel et al., 2014b). [top]
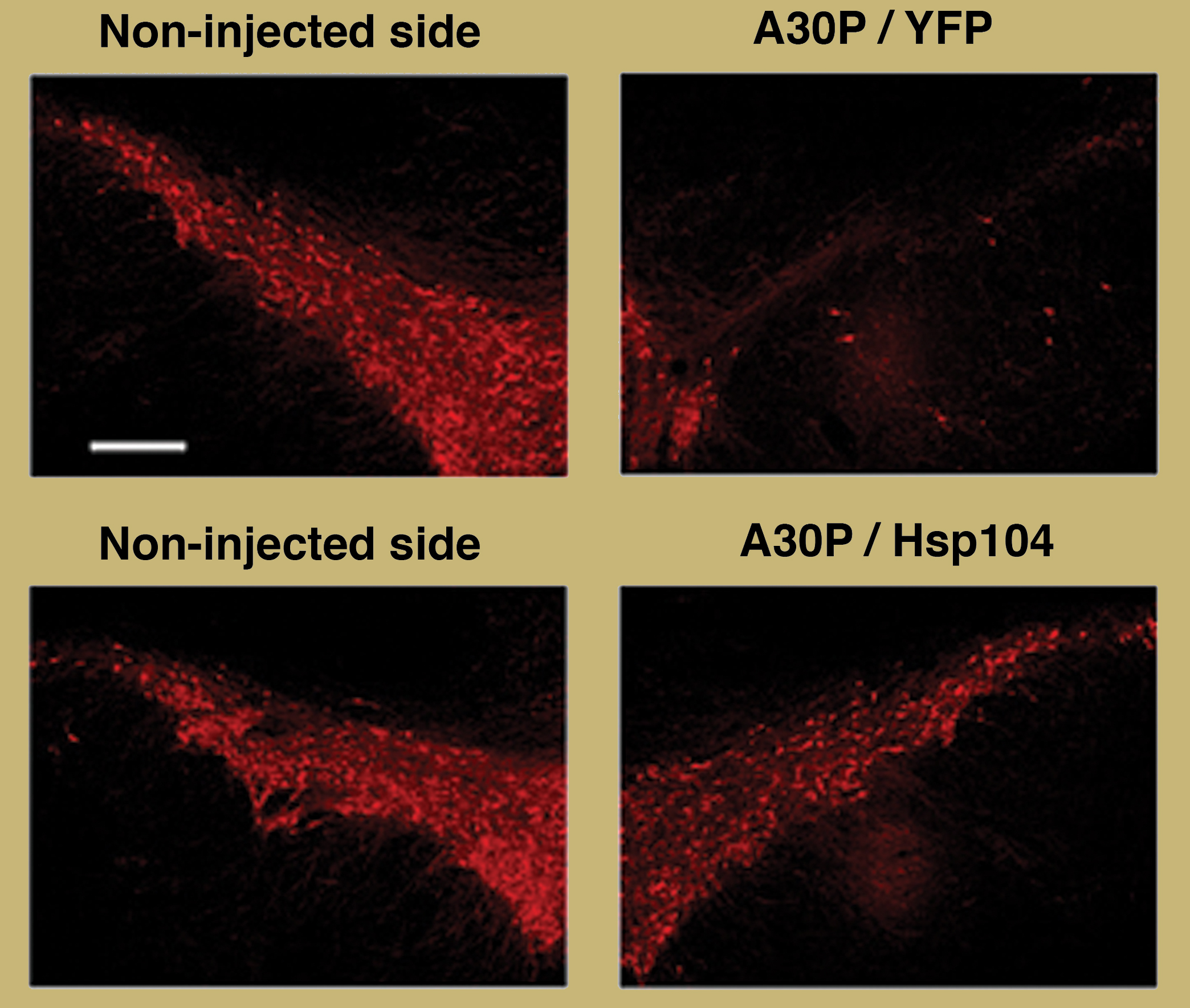
Hsp104 protects dopaminergic neurons (red) against alpha-synuclein toxicity in the substantia nigra of the rat brain (Lo Bianco et al., 2008).
3) Defining the metazoan disaggregase machinery. The loss of Hsp104 from metazoan lineages is abrupt. The choanoflagellate protist, Monosiga brevicollis, one of the most advanced pre-metazoans has a clear Hsp104 homologue, whereas even early branching metazoans like the sea anemone, Nematostella vectensis do not. The reason underlying the loss of Hsp104 is unclear, especially because Hsp104 is well tolerated in animal systems. Whether mammals possess an analogous protein disaggregase (AAA+ protein or otherwise) has endured as an important open question. Recently, we have answered this question and identified a mammalian disaggregase system comprised of Hsp110, Hsp70 and Hsp40, which catalyzes the disaggregation and reactivation of chemically and thermally denatured aggregates, but is unable to rapidly remodel amyloid (Shorter, 2011; Torrente & Shorter, 2013). However, together with small heat shock proteins, Hsp110, Hsp70 and Hsp40 can slowly depolymerize amyloid fibers from their ends (Duennwald et al., 2012; Torrente & Shorter, 2013). We are now delineating the mechanism of Hsp110, Hsp70 and Hsp40 action . We are also interested in identifying additional metazoan disaggregases and novel chaperone systems (Guo et al., 2014). [top]
4) Defining how small molecules modulate amyloid folding trajectories. We employ a variety of small molecules as mechanistic probes to understand amyloid foldings pathways of Sup35 and Aβ42. These include 4,5-dianilinophthalimide (DAPH-1) and analogs, which dissolve Aβ42 fibers (that occur in Alzheimer's disease) and eliminate their neurotoxicity (Wang et al., 2008). DAPH-1 also disrupts Sup35 prion structure and function (Wang et al., 2008). We are interested in defining the mechanisms by which DAPH-1 and other small molecules modulate amyloid formation. Intriguingly, we have found that some small molecules select for the formation of drug-resistant prions or amyloids (Roberts et al., 2009; Shorter, 2010; Duennwald & Shorter, 2010). We have also discovered that specific small molecule combinations can preclude the formation of drug-resistant polymorphs or 'strains' (Roberts et al., 2009; Shorter, 2010; Duennwald & Shorter, 2010). We have designed specific aromatic foldamers that potently inhibit spontaneous and seeded Aβ42 and Aβ43 fibril assembly (Seither et al., 2014). Furthermore, we seek to elucidate synergies between small molecules and protein disaggregases that may accelerate the disruption of specific amyloid oligomers and fibers. Indeed, we also seek to identify small molecules that inhibit or enhance protein disaggregase activity (Torrente et al., 2014). [top]
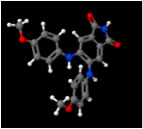
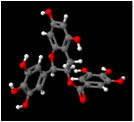
DAPH-12 (left) & EGCG (right) synergize to eliminate Sup35 prions (Roberts et al., 2009)
5) Defining the misfolding trajectories of RNA-binding proteins bearing prion-like domains in amyotrophic lateral sclerosis and other neurodegenerative disorders. Finally, we are investigating the mechanisms by which certain RNA-binding proteins, including TDP-43 and FUS, misfold and aggregate in various neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) with ubiquitin positive inclusions (FTLD-U) (Johnson et al., 2009; Sun et al., 2011); Qiu et al., 2014; Jackrel et al., 2014a; Jackrel & Shorter, 2014b). Intriguingly, using a bioinformaics approach we have discovered that TDP-43 and FUS harbor a prion-like domain similar to the domain in Sup35, Ure2 and other yeast prion proteins that confers prionogenicity (Cushman et al., 2010; Sun et al., 2011; Gitler and Shorter, 2011; King et al., 2012; Li et al., 2013). Remarkably, our bioinformatics approach indicates that several RNA-binding proteins in the human genome harbor prion-like domains similar to FUS and TDP-43 (Gitler and Shorter, 2011; Couthouis et al., 2011; Couthouis et al., 2012; King et al., 2012; Li et al., 2013; Kim et al., 2013; Shorter & Taylor, 2013). We have determined that two of these, TAF15 and EWSR1, are intrinsically aggregation prone and connected through pathology and genetics to ALS (Couthouis et al., 2011; Couthouis et al., 2012). We are interested to determine whether other RNA-binding proteins bearing prion-like domains misfold and contribute to other neurodegenerative disorders (King et al., 2012; Li et al., 2013). Indeed, we recently established that mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and amyotrophic lateral sclerosis (Kim et al., 2013; Shorter & Taylor, 2013). We aim to understand how the prion-like domain enables misfolding and whether these RNA-binding proteins access prion-like conformers. We are also elucidating methods to prevent or reverse the misfolding of various RNA-binding proteins with prion-like domains and mitigate their toxicity (Sun et al., 2011; Armakola et al., 2012; Jackrel et al., 2014a; Jackrel & Shorter, 2014b). [top]
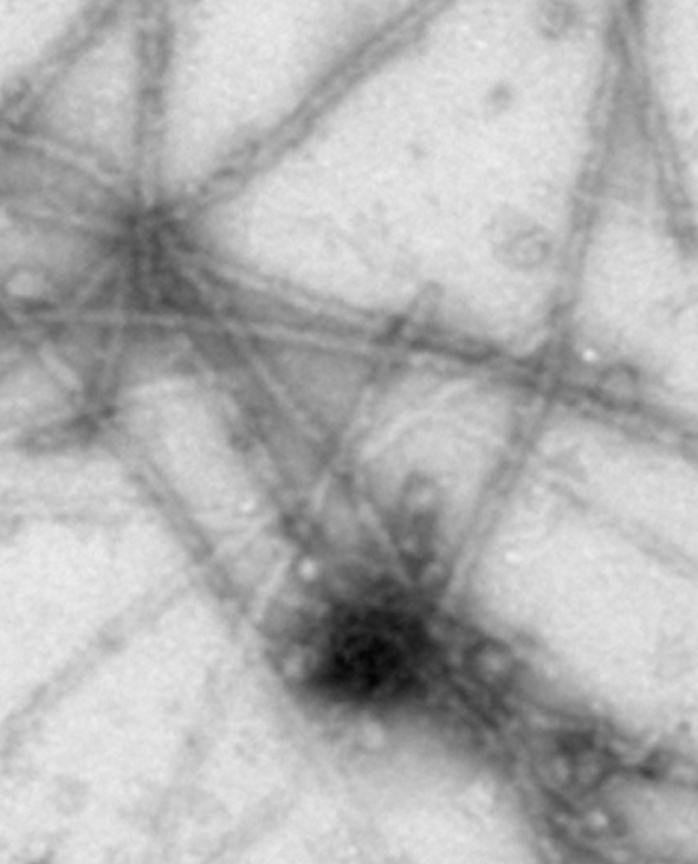
Pure FUS 1-422 forms linear polymers (Sun et al., 2011)
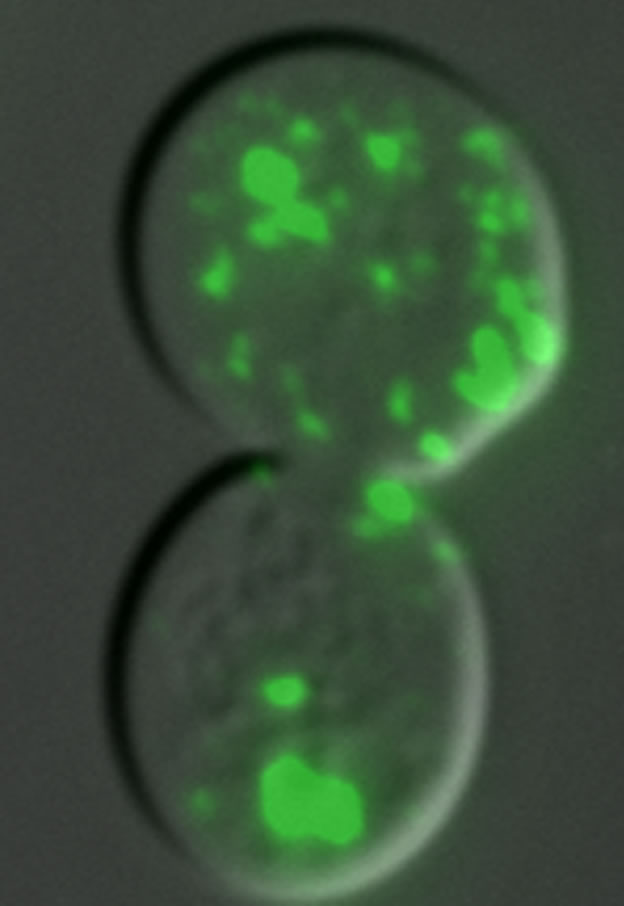
FUS-YFP aggregates in the yeast cytoplasm (Sun et al, 2011).
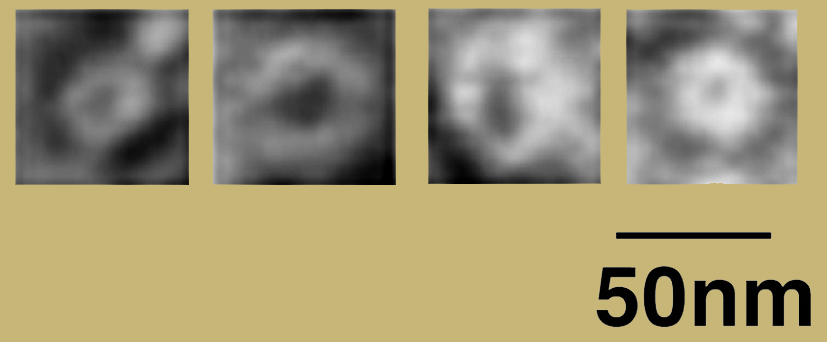
Oligomeric forms of TDP-43 (Johnson et al., 2009).
[top]







