The tumors of Neurofibromatosis
Neurofibromatosis is a genetic disorder that causes tumors predominantly in the Schwann cells of the peripheral nervous system (Bhatheja & Field, 2006). The sites of tumors include the skin, the spinal cord, and the brain. Although most of these tumors are benign, some can become malignant- the most common malignant tumors being malignant peripheral nerve sheath tumors (MPNSTs). There are three recognized types of neurofibromatosis: neurofibromatosis type 1 (NF1), neurofibromatosis type 2 (NF2), and schwannomatosis (SWN). Each type is caused by different mutations (Tamura, 2021).
NF1, also known is von Recklinghausen disease, is the most common type of neurofibromatosis. A mutation on the NF1 gene located on chromosome 17 results in loss of function or decreased expression of neurofibromin. Patients are born with one defective copy of NF1. When Schwann cells lose their remaining copy, they grow into to tumors. It is estimated every 1 in 3000 people have an NF1 mutation. Approximately 30-50% of cases are spontaneous mutation but if a parent carries the mutation, the child will have a 50% chance of inheriting the mutation. Symptoms of NF1 include café-au-lait spots, cutaneous neurofibromas, plexiform neurofibromas, neurological problems, and other complications.
The most common tumors are Neurofibromas. In 8-13% of NF1 patients neurofibromas will acquire additional mutations and develop int malignant peripheral nerve sheet tumors (MPNST). MPNST are associated with TP53 mutations as well as other genes. Some MPNST are sporadic and arise in patients without NF1. Current treatments for MPNST include surgical resection, radiation therapy, and chemotherapy (Somatilaka, Sadek, McKay, & Le, 2022).
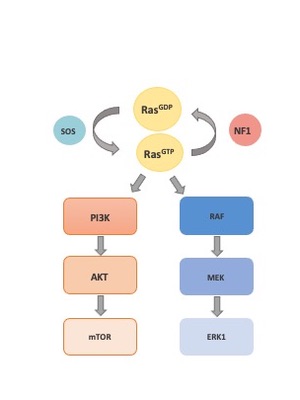
NF1 encodes the protein neurofibromin. Neurofibromin is a negative regulator of Ras, a Ras-GAP protein. Its loss leads to activation of the Ras/ Raf/MEK / ERK pathway. This can be seen in Pharmacomb analysis, as the NF1 cells are sensitive to MEK and ERK inhibitors. Selumetinib, a MEK inhibitor, is currently the only FDA approved targeted therapy for NF1, though it is approved only for neurofibromas.
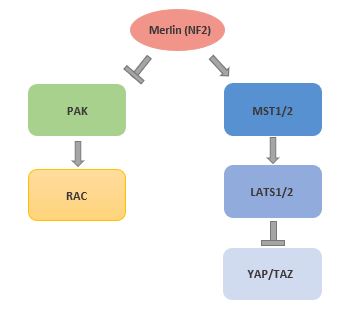
NF2 is less common than NF1 with an estimated 1 in 40,000 people possessing an NF2 mutation. The NF2 gene is located on chromosome 22 and produces the gene product merlin, a cytoskeletal tumor suppressor protein. As with NF1, NF2 is inherited, and tumors arise when the Schwann cells lose their one functional copy. NF2 tumors are more localized in the central and peripheral nervous system. Symptoms include schwannomas, meningiomas, ependymomas, visual problems, and other complications. NF2 binds and inhibits Pak kinases and also inhibits the Hippo pathway. Hence Pak and Hippo inhibitors have been implicated as targets, though there are no approved drugs for NF2 Schwannomas.
SWN is the rarest condition of the three types of neurofibromatosis and affect less than 1 in 40,000 people. Mutation of SMARCB or LZTR1 genes are often associated with SWN and there is around a 15% chance of inheriting the disease. Symptoms of SWN overlap with NF2, however, one third of cases have schwannomatosis features limited to only one part of the body. We do not have cell lines from SWN tumors.
Further reading:
Bhatheja, K., & Field, J. (2006). Schwann cells: Origins and role in axonal maintenance and regeneration. The International Journal of Biochemistry & Cell Biology, 38(12), 1995-1999
Tamura, R. (2021). Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis. Int J Mol Sci, 22(11),PMC8198724, doi:10.3390/ijms22115850
Somatilaka, B. N., Sadek, A., McKay, R. M., & Le, L. Q. (2022). Malignant peripheral nerve sheath tumor: models, biology, and translation. Oncogene, 41(17), 2405-2421,PMC9035132, doi:10.1038/s41388-022-02290-1
