Lab Projects
Theme 1: Locking and Unlocking Cardiac Maturation
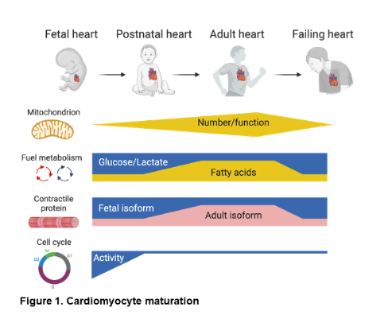 What is cardiac maturation? Early heart development involves a cascade of signaling and transcriptional events that drive cardiomyocyte (CM) lineage determination, proliferation, and organ morphogenesis leading to the formation of the cardiac chambers and great vessels. Much less is known about the developmental maturation process which begins in late fetal stages and proceeds into the postnatal period, resulting in the terminally differentiated adult heart. In contrast to early development, this process involves coordinated metabolic, structural, and contractile maturation of the post-mitotic CM (Figure 1). This terminal maturation process is critical for the development and maintenance of a fully functional adult mammalian heart.
What is cardiac maturation? Early heart development involves a cascade of signaling and transcriptional events that drive cardiomyocyte (CM) lineage determination, proliferation, and organ morphogenesis leading to the formation of the cardiac chambers and great vessels. Much less is known about the developmental maturation process which begins in late fetal stages and proceeds into the postnatal period, resulting in the terminally differentiated adult heart. In contrast to early development, this process involves coordinated metabolic, structural, and contractile maturation of the post-mitotic CM (Figure 1). This terminal maturation process is critical for the development and maintenance of a fully functional adult mammalian heart.
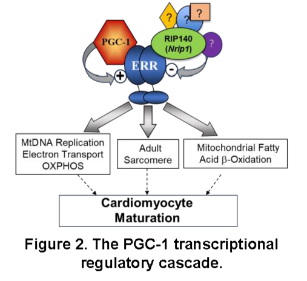 Over the past decade, studies in the Kelly lab defined a transcriptional regulatory circuitry that drives postnatal mitochondrial biogenesis and maturation in heart and skeletal muscle.1,2 We found that members of the nuclear receptor superfamily, the peroxisome proliferator-activated receptors (PPARs) and the estrogen-related receptors (ERRs), drive the high expression of genes involved in fatty acid oxidation (FAO), oxidative phosphorylation (OXPHOS), and other mitochondrial energy transduction pathways in the postnatal heart (Figure 2).3-12 We next discovered that full activation of PPARs and ERRs require the action of the inducible transcriptional coregulators, PPARg coactivator 1 (PGC-1) a and b (Figure 2).13,14 Importantly, this transcriptional regulatory circuitry is partially de-activated in pathological cardiac hypertrophy and heart failure (HF), resulting in a dynamic shift towards a more fetal-like metabolic state that likely contributes to the “energy starvation” of the failing heart.15-21 A key question remained: How is postnatal maturation of the CM mitochondrial machinery matched with energy-utilizing processes such as growth and contractile function? In other words, how are the mitochondrial and structural components of the developing and adult heart coordinately regulated? New insight was gained by the results of our recent studies showing that the largely functionally redundant nuclear receptors, ERRa and g (Figure 2), serve a central role in CM differentiation and maturation.22-25 Prenatal cardiac-specific ERRa/g knock-out (KO) in mice resulted in postnatal cardiomyopathy (CMP) with features of ventricular non-compaction and immature mitochondria.22 Conditional postnatal targeting of cardiac ERRa/g in mice, and in differentiating human iPSC-CM, demonstrated that ERRa and g are necessary for both energy metabolic and contractile protein perinatal fetal-adult gene switches.22,23 These results demonstrated that PGC-1/ERR signaling is critical for the coordinate regulation of mitochondrial ATP production and energy-consuming contractile processes during cardiac maturation and in the adult heart. This conceptual breakthrough forms the foundation for the projects in Theme 1.
Over the past decade, studies in the Kelly lab defined a transcriptional regulatory circuitry that drives postnatal mitochondrial biogenesis and maturation in heart and skeletal muscle.1,2 We found that members of the nuclear receptor superfamily, the peroxisome proliferator-activated receptors (PPARs) and the estrogen-related receptors (ERRs), drive the high expression of genes involved in fatty acid oxidation (FAO), oxidative phosphorylation (OXPHOS), and other mitochondrial energy transduction pathways in the postnatal heart (Figure 2).3-12 We next discovered that full activation of PPARs and ERRs require the action of the inducible transcriptional coregulators, PPARg coactivator 1 (PGC-1) a and b (Figure 2).13,14 Importantly, this transcriptional regulatory circuitry is partially de-activated in pathological cardiac hypertrophy and heart failure (HF), resulting in a dynamic shift towards a more fetal-like metabolic state that likely contributes to the “energy starvation” of the failing heart.15-21 A key question remained: How is postnatal maturation of the CM mitochondrial machinery matched with energy-utilizing processes such as growth and contractile function? In other words, how are the mitochondrial and structural components of the developing and adult heart coordinately regulated? New insight was gained by the results of our recent studies showing that the largely functionally redundant nuclear receptors, ERRa and g (Figure 2), serve a central role in CM differentiation and maturation.22-25 Prenatal cardiac-specific ERRa/g knock-out (KO) in mice resulted in postnatal cardiomyopathy (CMP) with features of ventricular non-compaction and immature mitochondria.22 Conditional postnatal targeting of cardiac ERRa/g in mice, and in differentiating human iPSC-CM, demonstrated that ERRa and g are necessary for both energy metabolic and contractile protein perinatal fetal-adult gene switches.22,23 These results demonstrated that PGC-1/ERR signaling is critical for the coordinate regulation of mitochondrial ATP production and energy-consuming contractile processes during cardiac maturation and in the adult heart. This conceptual breakthrough forms the foundation for the projects in Theme 1.
Why is understanding cardiac maturation important? Deciphering the regulatory mechanisms involved in cardiac maturation has important translational implications. Early during the development of HF, the CM reverts to a more immature or “fetal-like” state (Figure 1). Whereas this fetal shift may represent an evolved adaptation to acute stress, over time, constrained energetics and contractile function related to the less-differentiated state contribute to the pathogenesis of HF. Secondly, impaired cardiac maturation likely contributes to congenital forms of heart disease and genetic CMPs. Third, early attempts at replacing dead myocytes with stem-cell derived CMs have been hindered by lack of full maturation. Lastly, transient reversion of the CM to a more fetal-like state may enable cell cycle re-entry and, thus, cardiac regeneration. Therefore, strategies that target CM maturation may open exciting new therapeutic avenues for heart disease.
Project 1. Establish an annotated human cardiomyocyte ERRa/g genomic binding site catalog and delineate its regulation during developmental maturation and in the failing heart. In this project, we seek to establish a human genomic catalog and topologic blueprint for binding sites occupied by ERRg, ERRa, and their coregulator RIP140 using genomic techniques such as “cut & run” and RNA sequencing. We will assess how this genomic landscape is modified in the developing and failing mouse and human heart. The functional importance of a subset of these regulatory elements will be determined including mutational perturbation of the elements via CRISPR-Cas gene editing in human iPSC-CMs.
Project 2. Identify non-coding ERR DNA binding site variants that may contribute to human heart disease. It has been estimated that 50% or more of congenital heart disease (CHD) is due to mutations in non-coding regions of the genome. This is a new frontier in human genetics. The central hypothesis for this aim is that disturbances in CM differentiation and maturation contribute to the development and phenotypic expression of some forms of human congenital heart disease (CHD) and cardiomyopathy (CMP). And that genetic variants that alter ERRa/g DNA binding at regulatory sites that control cardiac-specific processes such as contractile function and developmental programs, contribute to the development of CHD and CMP. Common and rare non-coding DNA variants in human CM ERR binding sites are being identified and characterized by probing well-phenotyped human heart disease cohorts with the assistance of the human genomic catalog. Selected variants will be further pursued as disease-causing or disease-modifying.
20efProject 3. Determine the utility of maintaining or re-activating the cardiac maturation process in the failing heart. The CM ERR genomic landscape is regulated during postnatal CM maturation and reverts towards a maladaptive fetal pattern in the failing heart. We hypothesize that this reversion to an immature state is maladaptive and contributes to heart failure. Therefore, we propose that postnatal targeting of components of the ERR complex (Figure 2) to reactivate or “lock-in” CM maturation by inhibiting RIP140 or activating ERR signaling to maintain or re-activate the CM maturation program will protect against the development of HF. This will be accomplished by re-activating the CM maturation transcriptional regulatory circuitry in mice with HF and in an engineered human heart tissue (EHT) platform.
Project 4. Unlocking metabolic maturation to enable CM proliferation. Evidence is emerging that the postnatal CM can re-enter the cell cycle - a “holy grail” of cardiovascular research.26-29 During the fetal and early postnatal periods when the CM is still in an immature state, CM proliferation occurs as has been shown by response of the neonatal heart to injury.30-32 Following this period and in the adult, CMs exit the cell cycle. With cellular proliferation, a shift to aerobic glycolysis occurs as described by Warburg in classic studies of cancer cells.33 Interestingly, recent evidence suggests that this metabolic re-programming may serve as a driver of CM proliferation. Consistent with this notion, a recent study demonstrated that CMs in mice deficient for CPT1b, a rate-limiting enzyme in fatty acid oxidation, were capable of proliferation in the context of ischemic injury.34 The mechanisms involved in the re-entry of CMs into the cell cycle by re-programming fuel utilization are unknown but some evidence suggests crosstalk between intermediary metabolites and the epigenome.34 These results raise the intriguing possibility that transient “unlocking” of the CM fuel metabolic maturation program will enable CM proliferation, particularly in the context of an injurious stressor such as ischemia. We hypothesize that a shift to a more immature glycolytic state with deactivation of the PGC-1 cascade will enhance CM proliferation in the context of injury. This project involves deactivating selected components of the PGC-1/ERR/PPAR regulatory circuitry to enable CM proliferation during neonatal development and in the injured heart.
Theme 2. Muscle Metabolic Fitness in Health and Disease
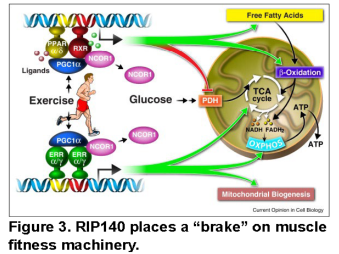 Muscle fitness is altered in a myriad of primary muscle disorders and secondary to chronic diseases such as heart disease, cancer, and in sarcopenic states caused by aging. The PGC-1 regulatory circuitry has been shown to mediate the benefits of exercise to muscle metabolic endurance fitness (Figure 3). We seek to harness components of this transcriptional regulatory circuit to enhance muscle fitness.
Muscle fitness is altered in a myriad of primary muscle disorders and secondary to chronic diseases such as heart disease, cancer, and in sarcopenic states caused by aging. The PGC-1 regulatory circuitry has been shown to mediate the benefits of exercise to muscle metabolic endurance fitness (Figure 3). We seek to harness components of this transcriptional regulatory circuit to enhance muscle fitness.
Project 1. Probing the role of RIP140 in muscle fitness and harnessing its functions in disease states. This project has evolved based on an interesting phenotype of muscle specific PPAR (MCK-PPARd) transgenic mice generated in our laboratory.35,36 MCK-PPARd mice exhibit an endurance trained phenotype and have been referred to as “marathon mice”. More recently, we have targeted an endogenous repressor of PPARs and ERRs termed RIP140 (Figure 2) in muscle. Muscle RIP140 KO mice exhibit a remarkable endurance phenotype. The RIP140-deficient muscle exhibits increased mitochondrial density, a shift toward oxidative fibers, angiogenesis, and adaptive remodeling of neuromuscular junctions in the absence of exercise training. Moreover, the RIP140 KO mice can train to higher levels of VO2max compared to wild-type littermates. This project involves defining the array of RIP140 target genes and corresponding transcription factor effectors that are responsible for this amazing phenotype. In addition, RIP140 KO mice will be used to determine if inhibiting this muscle transcriptional co-regulator ameliorates muscle diseases such as muscular dystrophy, muscle injury, and sarcopenia.
BIBLIOGRAPHY
1. Kelly DP and Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357-68.
2. Kelly DP and Scarpulla RC. Transcriptional control of striated muscle mitochondrial biogenesis and function. In: J. A. Hill and E. N. Olson, eds. Muscle: Fundamental Biology and Mechanisms of Disease. 1st ed. London: Elsevier Academic Press; 2012: 203-215.
3. Carter ME, Gulick T, Moore DD and Kelly DP. A pleiotropic element in the medium-chain acyl coenzyme A dehydrogenase gene promoter mediates transcriptional regulation by multiple nuclear receptor transcription factors and defines novel receptor-DNA binding motifs. Mol Cell Biol. 1994;14:4360-72.
4. Gulick T, Cresci S, Caira T, Moore DD and Kelly DP. The peroxisome proliferator-activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression. Proc Natl Acad Sci USA. 1994;91:11012-6.
5. Leone TC, Cresci S, Carter ME, Zhang Z, Lala DS, Strauss AW and Kelly DP. The human medium chain acyl-CoA dehydrogenase gene promoter consists of a complex arrangement of nuclear receptor response elements and Sp1 binding sites. J Biol Chem. 1995;270:24622.
6. Disch DL, Rader TA, Cresci S, Leone TC, Barger PM, Vega R, Wood PA and Kelly DP. Transcriptional control of a nuclear gene encoding a mitochondrial fatty acid oxidation enzyme in transgenic mice: role for nuclear receptors in cardiac and brown adipose expression. Mol Cell Biol. 1996;16:4043-51.
7. Brandt JM, Djouadi F and Kelly DP. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor alpha. The Journal of biological chemistry. 1998;273:23786-92.
8. Djouadi F, Weinheimer CJ, Saffitz JE, Pitchford C, Bastin J, Gonzalez FJ and Kelly DP. A gender-related defect in lipid metabolism and glucose homeostasis in peroxisome proliferator- activated receptor alpha- deficient mice. J Clin Invest. 1998;102:1083-91.
9. Leone TC, Weinheimer CJ and Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci USA. 1999;96:7473-8.
10. Campbell FM, Kozak R, Wagner A, Altarejos JY, Dyck JR, Belke DD, Severson DL, Kelly DP and Lopaschuk GD. A role for peroxisome proliferator-activated receptor alpha (PPARalpha ) in the control of cardiac malonyl-CoA levels: reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J Biol Chem. 2002;277:4098-103.
11. Huss JM, Torra IP, Staels B, Giguere V and Kelly DP. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol. 2004;24:9079-91.
12. Burkart EM, Sambandam N, Han X, Gross RW, Courtois M, Gierasch CM, Shoghi K, Welch MJ and Kelly DP. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. The Journal of clinical investigation. 2007;117:3930-9.
13. Puigserver P, Wu Z, Park CW, Graves R, Wright M and Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829-39.
209b14. Lin J, Puigserver P, Donovan J, Tarr P and Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta ), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem. 2002;277:1645-8.
15. Sack MN, Rader TA, Park S, Bastin J, McCune SA and Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94:2837-42.
16. Sack MN, Disch DL, Rockman HA and Kelly DP. A role for Sp and nuclear receptor transcription factors in a cardiac hypertrophic growth program. Proc Natl Acad Sci USA. 1997;94:6438-43.
17. Barger PM, Brandt JM, Leone TC, Weinheimer CJ and Kelly DP. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin Invest. 2000;105:1723-30.
18. Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M and Giguere V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab. 2007;5:345-56.
19. Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, Giguere V, Murphy E and Kelly DP. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007;6:25-37.
20. Lehman JJ, Boudina S, Banke NH, Sambandam N, Han X, Young DM, Leone TC, Gross RW, Lewandowski ED, Abel ED and Kelly DP. The transcriptional coactivator PGC-1alpha is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. American journal of physiology Heart and circulatory physiology. 2008;295:H185-96.
21. Vega RB and Kelly DP. Cardiac nuclear receptors: architects of mitochondrial structure and function. J Clin Invest. 2017;127:1155-1164.
22. Sakamoto T, Matsuura TR, Wan S, Ryba DM, Kim JU, Won KJ, Lai L, Petucci C, Petrenko N, Musunuru K, Vega RB and Kelly DP. A Critical Role for Estrogen-Related Receptor Signaling in Cardiac Maturation. Circ Res. 2020;126:1685-1702.
23. Sakamoto T, Batmanov K, Wan S, Guo Y, Lai L, Vega RB and Kelly DP. The nuclear receptor ERR cooperates with the cardiogenic factor GATA4 to orchestrate cardiomyocyte maturation. Nat Commun. 2022;13:1991.
24. Cao Y, Zhang X, Akerberg BN, Yuan H, Sakamoto T, Xiao F, VanDusen NJ, Zhou P, Sweat ME, Wang Y, Prondzynski M, Chen J, Zhang Y, Wang P, Kelly DP and Pu WT. In Vivo Dissection of Chamber-Selective Enhancers Reveals Estrogen-Related Receptor as a Regulator of Ventricular Cardiomyocyte Identity. Circulation. 2023;147:881-896.
25. Sakamoto T and Kelly DP. Cardiac Maturation. J Mol Cell Cardiol. 2024;187:38-50.
26. Heallen T, Morikawa Y, Leach J, Tao G, Willerson JT, Johnson RL and Martin JF. Hippo signaling impedes adult heart regeneration. Development. 2013;140:4683-90.
27. Monroe TO, Hill MC, Morikawa Y, Leach JP, Heallen T, Cao S, Krijger PHL, de Laat W, Wehrens XHT, Rodney GG and Martin JF. YAP Partially Reprograms Chromatin Accessibility to Directly Induce Adult Cardiogenesis In Vivo. Dev Cell. 2019;48:765-779 e7.
28. Leach JP, Heallen T, Zhang M, Rahmani M, Morikawa Y, Hill MC, Segura A, Willerson JT and Martin JF. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature. 2017;550:260-264.
29. Liu S, Li K, Wagner Florencio L, Tang L, Heallen TR, Leach JP, Wang Y, Grisanti F, Willerson JT, Perin EC, Zhang S and Martin JF. Gene therapy knockdown of Hippo signaling induces cardiomyocyte renewal in pigs after myocardial infarction. Sci Transl Med. 2021;13.
30. Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah AM, Zhang H, Faber JE, Kinter MT, Szweda LI, Xing C, Hu Z, Deberardinis RJ, Schiattarella G, Hill JA, Oz O, Lu Z, Zhang CC, Kimura W and Sadek HA. Hypoxia induces heart regeneration in adult mice. Nature. 2017;541:222-227.
31. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN and Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078-80.
32. Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Aroumougame A, Shah AM, Szweda LI and Sadek HA. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565-79.
33. Warburg O and Minami S. Versuche an Überlebendem Carcinom-gewebe. Klin Wochenschr. 1923;2:776-777.
34. Li X, Wu F, Gunther S, Looso M, Kuenne C, Zhang T, Wiesnet M, Klatt S, Zukunft S, Fleming I, Poschet G, Wietelmann A, Atzberger A, Potente M, Yuan X and Braun T. Inhibition of fatty acid oxidation enables heart regeneration in adult mice. Nature. 2023;622:619-626.
35. Gan Z, Burkart-Hartman EM, Han DH, Finck B, Leone TC, Smith EY, Ayala JE, Holloszy J and Kelly DP. The nuclear receptor PPARbeta/delta programs muscle glucose metabolism in cooperation with AMPK and MEF2. Genes Dev. 2011;25:2619-30.
36. Gan Z, Rumsey J, Hazen BC, Lai L, Leone TC, Vega RB, Xie H, Conley KE, Auwerx J, Smith SR, Olson EN, Kralli A and Kelly DP. Nuclear receptor/microRNA circuitry links muscle fiber type to energy metabolism. J Clin Invest. 2013;123:2564-75.