|
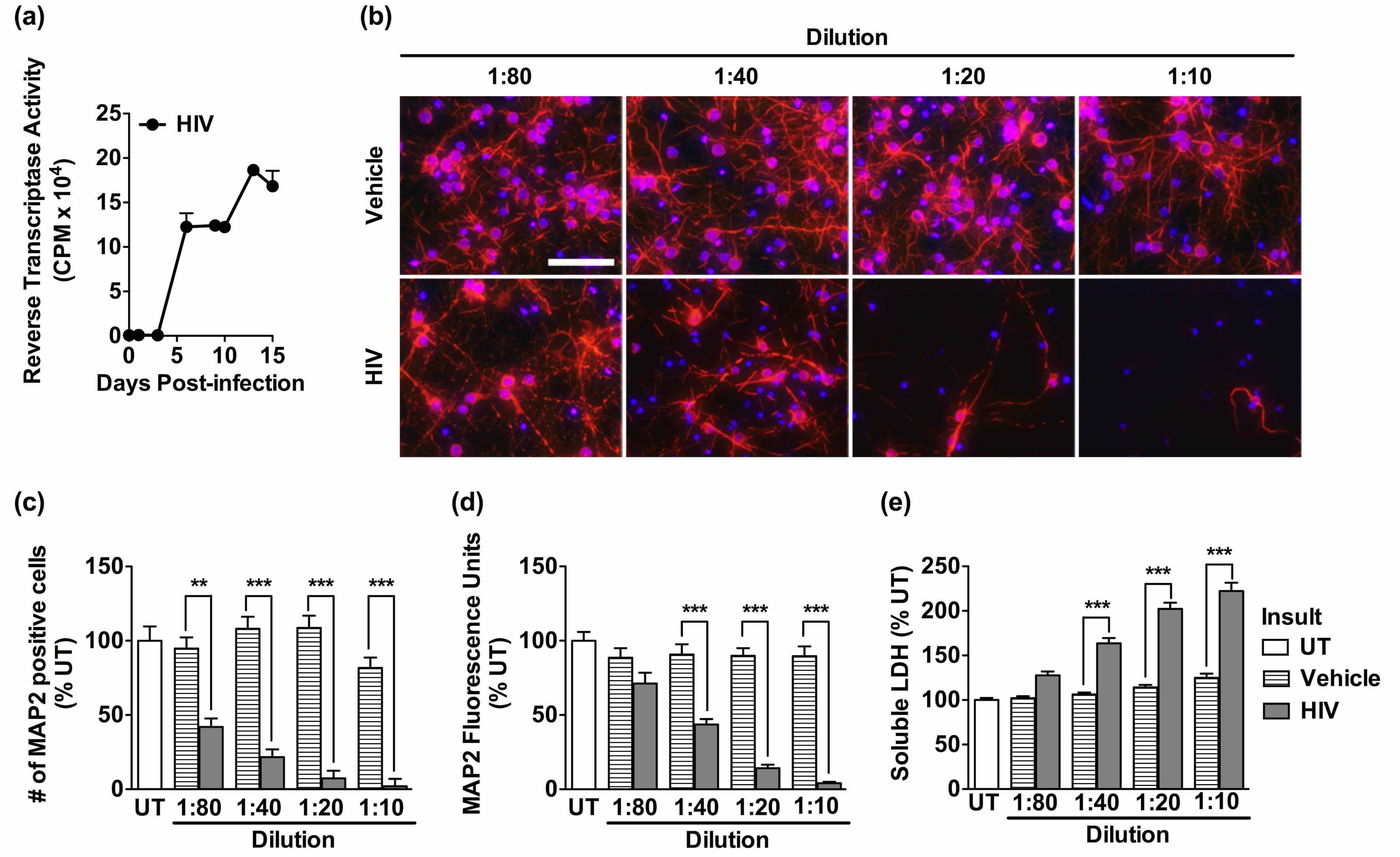
We hypothesized that cellular adaptive stress responses, which can be triggered by virus infection, are not only activated in HIV monocyte-derived macrophages (HIV/MDM) but also modify neurotoxin release and inflammatory cascades associated with neurodegeneration. We have developed an in vitro model of HIV neurotoxicity utilizing human HIV/MDM and primary rat cortical neurons.
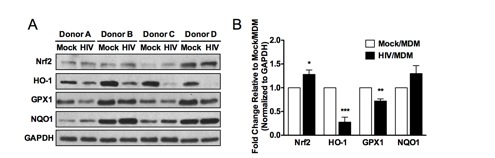
With this system, we have demonstrated that HIV infection of MDM results in the modulation of specific components of the antioxidant response and the integrated stress response (ISR). Our results suggest that pharmacological agents that activate the antioxidant response have therapeutic potential for HAND, through suppression of HIV replication and neurotoxin production in HIV/MDM. Our ongoing studies are defining the roles for the antioxidant response in suppressing inflammatory mediators and neurotoxin production by HIV/MDM. Understanding how cellular adaptive stress responses respond to HIV infection and the consequences of this activation on HIV replication and neurotoxin production will improve our understanding of HAND pathogenesis and provide insights for therapeutic strategies.
Induction of the antioxidant response element (ARE) reduces HIV/MDM neurotoxicity and attenuates HIV replication in macrophages
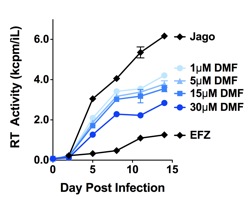
HIV infection and progression is associated with alterations in the oxidative state of infected cells. Oxidative stress can drive HIV replication through induction of NF-κB dependent mechanisms and through the up-regulation of inflammatory cytokines. Therefore, induction of the antioxidant response or suppression of inflammatory responses in macrophages could reduce HIV replication and neuronal damage following HIV infection of the CNS. A class of compounds known as fumarates, which include dimethyl fumarate (DMF) and monomethyl fumarate (MMF), have been shown to have anti-inflammatory activity and induce the antioxidant response in leukocytes, endothelial cells and mixed glial cell cultures. Formulations of these fumaric acid esters (FAE) have clinical efficacy for the autoimmune inflammatory disease psoriasis and recent clinical trials are examining FAE in phase 3 clinical trials for multiple sclerosis in the United States. While the precise molecular mechanism of DMF is not understood, it is known to inhibit the expression of inflammatory mediators (e.g. IL1β, IL-6, TNFα, and nitric oxide) and is a potent activator of the Nrf2-dependent anti-oxidant response element (ARE). Because of the proposed mechanism(s) of action of fumarates in the CNS, it is a potential therapeutic for limiting HIV-associated neurodegeneration. We have found the MMF/DMF can attenuate HIV replication in macrophages while simultaneously decreasing neurotoxin production by HIV/MDM. MMF/DMF are attractive therapeutic candidates for the treatment of HAND as they have antiviral and anti-inflammatory effects that target multiple steps in HAND pathogenesis.
Activation of the integrated stress response attenuates HIV replication in macrophages but enhances neurotoxin production
Accumulation of misfolded proteins in the endoplasmic reticulum (ER) lumen results in the activation of an adaptive response known as the ISR, formerly known as the unfolded protein response (UPR). The ISR is a quality control mechanism that can be activated during physiological stress (e.g., glucose deprivation, heat shock) and viral infection. The ISR reduces the load of misfolded proteins in the ER by attenuating new protein translation via eif2α and ISRegulating chaperones that mediate protein folding, such as BiP. During viral infection, the cell can experience an excess flux of viral proteins passing through the ER. Several viruses, including human cytomegalovirus and herpes simplex virus, have been shown capable of regulating cellular responses to infection, including the ISR, in order to promote cell survival during efficient viral replication. The effect of HIV infection on the ISR has not yet been investigated. We hypothesize that HIV infection induces and modulates the ISR in infected macrophages. We have demonstrated that HIV infection of human MDM results in a biphasic induction of phosphorylated-eif2α, with peaks of activation early after infection (24 hours) and during high replication periods. Phosphorylated-eif2α is a key indicator of cellular stress and directly attenuates protein translation and promotes molecular chaperones. We found that pharmacological induction of phosphorylated-eif2α attenuates HIV replication, demonstrating that these stress responses are part of an anti-viral cellular defense in the macrophage. However, induction of phosphorylated-eif2α enhances neurotoxin production in HIV/MDM, suggesting that the ISR can enhance neurotoxin production in HIV-infected macrophages.
|