Active Projects
Take a look at what we are working on.
Progression to type 2 diabetes (T2D) in individuals with insulin resistance depends on dysfunction and eventual loss of pancreatic β cells, resulting from chronic exposure to conditions associated with T2D, including elevated serum glucose and free fatty acids. These chronic stresses lead not only to β cell apoptosis but also to loss of cell identity due to disruption of the β cell gene expression program. Therefore, identifying factors that shape gene regulatory networks in β cells during stress is central to our understanding of T2D pathogenesis. RNA binding proteins (RBPs) shape gene expression programs by recognizing specific sequence elements and/or RNA secondary structures typically located in the untranslated regions (UTRs) of mRNAs, thereby allowing RBPs to regulate post-transcriptional processes, including splicing, translation, and RNA stability. mRNAs are bound by an array of RBPs throughout their life cycle, positioning RBPs to rapidly integrate cellular signals with changes in gene expression. The Stoffers laboratory recently identified the polyC binding protein (PCBP) family of RBPs as important players in the post-transcriptional regulatory landscape of pancreatic beta cells.
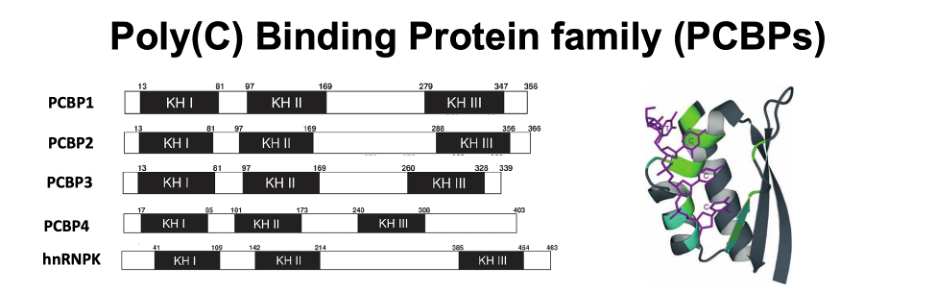
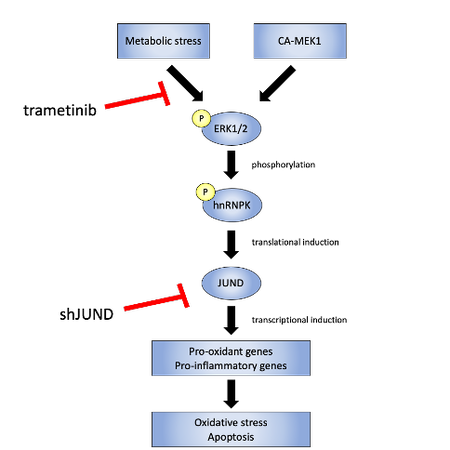
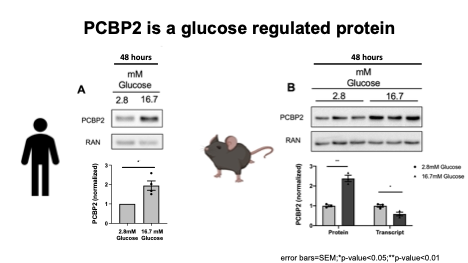
The Stoffers lab recently found that the PCBP hnRNPk translationally upregulates JUND in islets exposed to glucolipotoxicity conditions (high glucose and free fatty acid; GLT) and that depletion of JUND in β cells reduces oxidative stress and apoptosis caused by GLT. JUND is a transcription factor with anti-oxidant functions in other cell types but whose role in β cells was unknown. JunD was shown to regulate a cohort of genes that are commonly dysregulated during β cell dysfunction, including pro-oxidant and pro-inflammatory genes, consistent with this factor enhancing, rather than reducing, oxidative stress levels in β cells (Good et al, Molecular Metabolism 2019a) . Although hnRNPK level is unchanged, hnRNPK phosphorylation is altered during glucolipotoxicity. Finally, hnRNPK was found to regulate JUND in a DDX3X-dependent manner (Good et al, Molecular Metabolism 2019b) . This axis was found to be conserved in primary human islets, offering several new nodes for potential therapeutic intervention.
References:
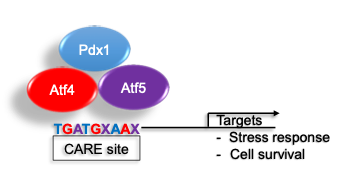
Loss of insulin secretion due to failure or death of the insulin secreting β cells is the central cause of diabetes. The cellular response to stress (endoplasmic reticulum (ER), oxidative, inflammatory) is essential to sustain normal β cell function and survival. We recently identified a novel PDX1 stress inducible complex (es) that regulates expression of stress and apoptosis genes to govern β cell survival.
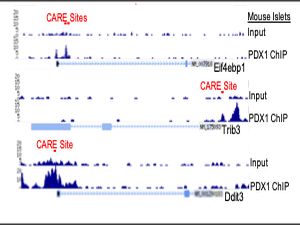

RNASeq identified a set of PDX1, ATF4 and ATF5 co-regulated genes enriched in stress and apoptosis functions. We further identified stress induced interactions among PDX1, ATF4, and ATF5. PDX1 chromatin occupancy peaks were identified over composite C/EBP-ATF (CARE) motifs of 26 genes; assessment of a subset of these genes revealed co-enrichment for ATF4 and ATF5. PDX1 occupancy over CARE motifs was conserved in the human orthologs of 9 of these genes. Of these, Glutamate Pyruvate Transaminase 2 (Gpt2), Cation transport regulator 1 (Chac1), and Solute Carrier Family 7 Member 1 (Slc7a1) induction by stress was conserved in human islets and abrogated by deficiency of Pdx1, Atf4, and Atf5 in Min6 cells.
Our results identify a novel PDX1 stress inducible complex (es) that regulates expression of stress and apoptosis genes to govern β cell survival.
References:
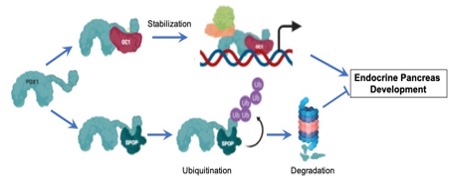
We hypothesize that PDX1/OC1 interactions, in part mediated by their IDPRs, regulate Pdx1 stability, cell cycle progression, and pancreatic endocrine differentiation during development.
The transcriptional networks critical for the proper development, differentiation, and expansion of beta cells, work through islet enhancers, super enhancers, and active promoters that form 3-dimensional hubs, some of which are coordinated by glucose. Pdx1 is a critical member of the beta cell transcriptional network, playing critical roles in early pancreas specification, regulation of organ size, and in beta cell formation, proliferation, identity, and function. Notably, Pdx1 is mutated in monogenic forms of human diabetes and mutations in the C-terminus are associated with human type 2 diabetes.
We previously found that truncation of the Pdx1 C-terminal domain in vivo impacts pancreas size and dramatically reduces the number of endocrine progenitors in mice, but the effects of human C-terminal mutations on Pdx1 function and beta cell development have not been elucidated. The C-terminus contains a highly intrinsically disordered protein region (IDPR) with unknown function (aa210-223). IDPRs, commonly found within transcription factors, lack fixed secondary structure and are amenable to phase separation, properties that promote protein-protein interactions and transcriptional hub formation at super enhancers necessary for coordinated gene regulation. IDPRs may promote access of neighboring regions to post-translational modifications. The Pdx1 C-terminus also contains an evolutionarily conserved motif (aa224-238) that mediates protein-protein interaction with the E3 Cul3 substrate adaptor SPOP/PCIF1 to promote proteasomal degradation of Pdx1 as well as a low-glucose-dependent phosphorylation site at Ser268 that is associated with Pdx1 destabilization. Importantly, our team has found that the Pdx1 C-terminus also mediates interaction with the one cut homeodomain transcription factor Oc1/Hnf6 in multipotent pancreatic progenitor cells (MPCs) to establish the endocrine pancreas gene program, with long-term impact on postnatal islet function and beta cell compensation in response to proliferative stimuli. Our unpublished data indicate that interactions of Pdx1 with SPOP and OC1 occur via the same conserved C-terminus motif. Based on our published observations and on our substantial preliminary data, we hypothesize that PDX1/OC1 interactions, in part mediated by their IDPRs, regulate Pdx1 stability, cell cycle progression, and pancreatic endocrine differentiation during development. Our current efforts are directed at (1) determining the mechanisms whereby Pdx1 and Oc1 cooperate to establish a chromatin landscape permissive for endocrine differentiation and proliferation, (2) Defining the roles of the Pdx1 and Oc1 IDPRs in protein-protein interaction and endocrine differentiation, and (3) Defining the molecular mechanisms by which the Pdx1 C-terminal domain regulates protein stability and function during pancreas organogenesis and endocrine differentiation.
This project is a highly collaborative effort with the Gannon laboratory at Vanderbilt University, the Soleimanpour laboratory at the University of Michigan, and the Gadue laboratory at the Children’s Hospital of Philadelphia (CHOP).
References:
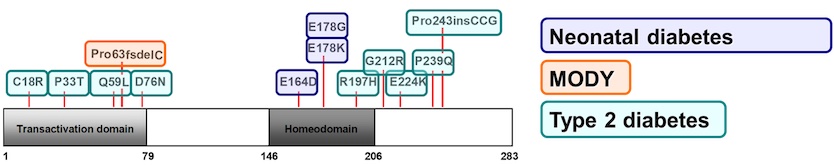
The Stoffers Lab is always ready to help our clinical and genetics colleagues by investigating the mechanisms by which human diabetes mutations influence PDX1 functions.
References:
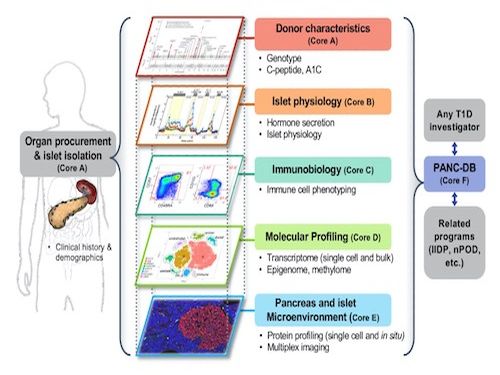
Dr. Stoffers directs the Penn Diabetes Research Center (DRC) Islet Cell Biology Core (ICBC). The objective of the Islet Cell Biology Core (ICBC) is to provide DRC members with state of the art support including experiment design, islet isolation, and performance of and/or training in an expansive range of assays for physiological and morphological assessment of pancreatic islet function and growth. We also work with investigators to adapt existing technologies to solve unique problems that cannot be addressed by standard methodologies.
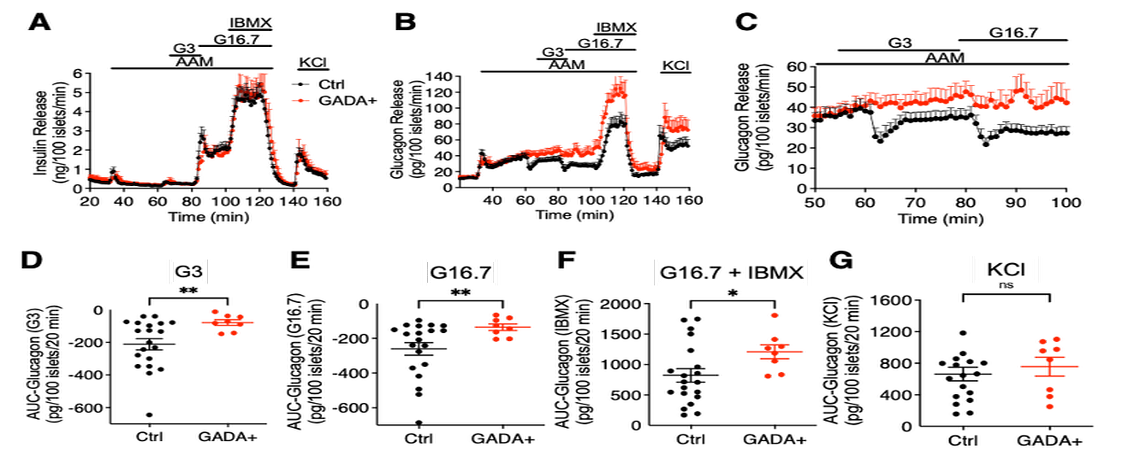
The ICBC plays a central role in the Human Pancreas Analysis Program efforts in both type 1 and type 2 diabetes through its key involvement in Islet Physiological Phenotyping of all HPAP donor islet preparations. Recent findings have highlighted the role of alpha cell defects in islet from GADA+ donors and in differences in function of islets from non-diabetic and diabetic donors of different ethnicities.
References:
Doliba NM, Rozo AV, Roman J, Qin W, Traum D, Gao L, Liu J, Manduchi E, Liu C, Golson ML, Vahedi G, Naji A, Matschinsky FM, Atkinson M, Powers AC, Brissova M, Kaestner KH, Stoffers DA for the HPAP Consortium. Alpha Cell Dysfunction in Islets from Non-Diabetic GADA+ Individuals. 2022, accepted but unpublished, Journal of Clinical Investigation.