Description of Research
Eukaryotic cells are compartmentalized into distinct membrane-bound organelles and vesicular structures, each with its own characteristic function and set of protein constituents. Work in our laboratory is focused on understanding how integral membrane protein complexes are assembled and sorted to the appropriate compartments within the late secretory and endocytic pathways, how sorting and assembly contribute to the biogenesis of specific organelles in several cell types, how these processes impact biological function in the pigmentary, blood clotting, and immune systems, and how they are thwarted by generally rare genetic diseases.
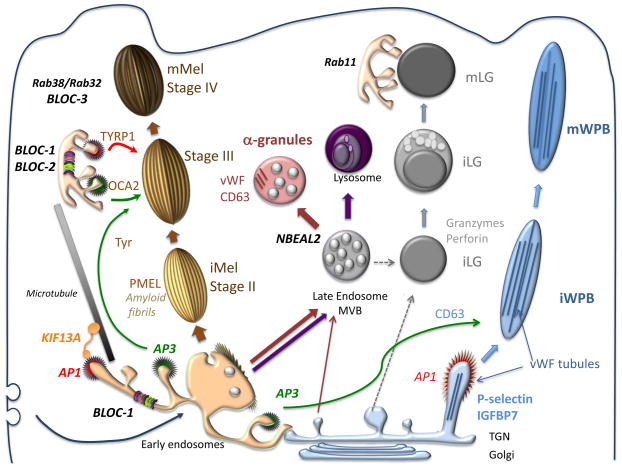
Model for Biogenesis and Docking of Four Vertebrate LROs1
Our primary focus over the past 20 years has been on melanosomes of pigmented cells. Melanosomes are unique lysosome-related organelles present only in cells that make melanin, the major synthesized pigment in mammals. Genetic defects in melanosome constituents or in their delivery to nascent melanosomes result in ocular or oculocutaneous albinism, characterized by lack of pigmentation in the eyes and or skin and concomitant visual impairment and susceptibility to skin and ocular cancers. Melanosomes are among a number of tissue-specific lysosome-related organelles that are malformed and dysfunctional in a group of rare heritable disorders, including Hermansky-Pudlak and Chediak-Higashi syndromes, and pigment cell-specific proteins that localize to melanosomes are targets for the immune system in patients with melanoma. In an effort to understand the molecular basis of these diseases, we are dissecting the molecular mechanisms that regulate how different stage melanosomes are formed and integrated with the endosomal pathway. We use biochemical, imaging, and genetic approaches to follow the fates of melanosome-specific and ubiquitous endosomal and lysosomal proteins within pigment cells from normal individuals or mice and disease models. Using these approaches, we are (1) outlining protein transport pathways that lead to the formation of these unusual organelles, (2) dissecting biochemical pathways that lead to their morphogenesis, and (3) defining how these processes are subverted by genetic disease. Current efforts focus on how factors that are deficient in patients and mouse models of the genetic disease, Hermansky-Pudlak syndrome, impact melanosome biogenesis. We are particularly interested in how these factors contribute to the formation and dynamics of tubular connections between endosomes and maturing melanosomes that facilitate cargo transport, as well as the formation of retrograde membrane carriers that retrieve unneeded proteins from melanosomes. We also have a number of projects focusing on how specific membrane constituents of melanosomes or lysosomes - specifically, transmembrane transporters - influence melanosome maturation. The genes encoding these transporters are either mutagenized in some albinism patients, vary in expression in individuals with distinct pigmentation levels, or both.
Because genetic diseases like Hermansky-Pudlak syndrome affect multiple organ systems, we study how similar sorting processes involved in melanosome biogenesis influence other organelles in different cell types. The first involves lysosome-related organelles in platelets called dense granules and alpha granules. When platelets are activated at sites of blood vessel damage, the contents of these granules are released, leading to optimal blood clot formation and platelet activation. Like melanosomes, dense granules are malformed in Hermansky-Pudlak syndrome, and in collaboration with the Poncz, Stalker and French laboratories at CHOP and Penn we are studying how dense granule contents are delivered within platelets and their precursors (megakaryocytes). Studies in collaboration with the Poncz and French labs also address the contents and secretion of alpha granules and their disruption in human bleeding disorders.
Another cellular system of interest is the dendritic cell, a master regulator of T cell-mediated immunity. Patients with Hermansky-Pudlak syndrome type 2 have recurrent bacterial infections, and we have found that this is at least in part due to defects in the way that dendritic cells sense bacterial infection. Normally, ingested bacteria trigger signaling by innate immune receptors present on the membrane enclosing the bacteria (the phagosome) or in the cytoplasm; this signaling is defective in dendritic cells from a mouse model of the disease due to impaired recruitment of the receptors and their signaling platforms to phagosomes or to silencing of cytoplasmic receptors by autophagy. Ongoing studies aim to dissect how phagosome membrane dynamics normally lead to signaling and how this is altered in disease states. Interestingly, preliminary data suggest that other innate immune cells in a different form of Hermansky-Pudlak syndrome may be hyperactive in the same signaling pathways, and we are trying to understand why together with the Behrens, Hamilton and Kelsen labs at CHOP.
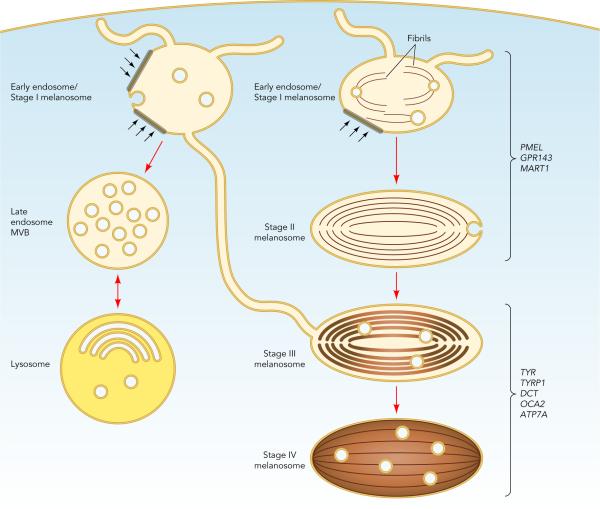
Melanosome Maturation and Segregation from Endocytic Organelles2
Finally, melanosome precursors in pigment cells harbor internal fibrils upon which melanins deposit in later stages. The main component of these fibrils is a pigment cell-specific protein, PMEL. Fibrils formed by PMEL in vitro display features common with amyloid formed in disease states such as Alzheimer and Parkinson diseases. By dissecting how PMEL forms amyloid under physiological conditions, we hope to determine how the formation of "good" and "bad" amyloid differs and thus how the formation of "bad" amyloid might be controlled.
References:
- Le, Linh, Julia Sirés-Campos, Graça Raposo, Cédric Delevoye, and Michael S Marks. “Melanosome Biogenesis in the Pigmentation of Mammalian Skin.” Integrative and Comparative Biology 61, no. 4 (October 1, 2021): 1517–45. https://doi.org/10.1093/icb/icab078.
- Bowman, Shanna L., Jing Bi-Karchin, Linh Le, and Michael S. Marks. “The Road to Lysosome-Related Organelles: Insights from Hermansky-Pudlak Syndrome and Other Rare Diseases.” Traffic 20, no. 6 (2019): 404–35. https://doi.org/10.1111/tra.12646.