Defining CD and Clinico-Pathology
Background:
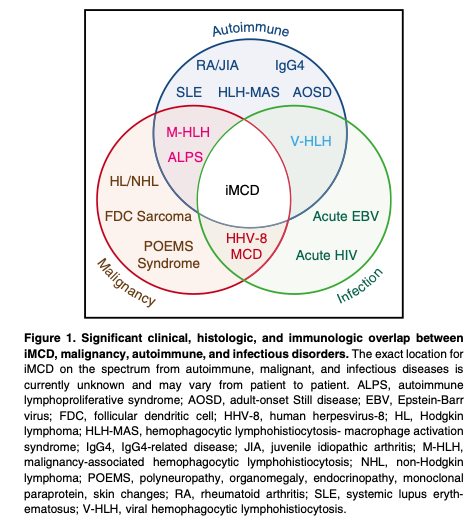
Castleman disease (CD) encompasses several clinicopathologic dis- orders at the intersection of hematology, oncology, rheumatology, and virology, with overlap in histopathologic and clinical features. His- torically, CD has been classified as unicentric or multicentric. A subset of multicentric CD (MCD) is caused by human herpesvirus-8 (HHV-8; also known as Kaposi sarcoma–associated herpesvirus) (HHV-8- associated MCD), whereas HHV-8–negative MCD cases remain idiopathic (iMCD). Unicentric CD (UCD) involves a single lymph node region showing characteristic “Castleman-like” histopathologic changes. Inflammatory manifestations are generally mild in UCD and usually disappear after surgical excision of the lymph node. In contrast, both iMCD and HHV-8–associated MCD are characterized by multifocal lymphadenopathy with a range of histopathology and episodic systemic inflammatory symptoms. HHV-8–associated MCD is most commonly diagnosed in HIV-infected or otherwise immunocompromised individuals. Virally- encoded interleukin (IL)-6 and human IL-6 are implicated in disease pathogenesis.
HHV-8–negative/iMCD is less well understood and has no specific biomarkers. Currently, iMCD is diagnosed when a constellation of nonspecific but characteristic lymph node histopathologic features commonly described as “hyaline vascular,” “plasmacytic,” or “mixed” are observed in patients with appropriate clinical features. HHV- 8–associated MCD and a range of malignant, autoimmune, and infectious disorders known to mimic these features (Figure 1) should be excluded. The etiology of iMCD is unknown, although it is hypothesized to involve one or more of the following mechanisms: autoimmunity/autoinflammation (ie, pathologic auto-antibodies or germline genomic alterations in inflammatory pathways); paraneoplastic (ie, somatic mutations in clonal cells); or infection with a virus other than HHV-8. It is possible that multiple pathways culminate in a cytokine storm that results in similar clinical presentations.
iMCD patients experience systemic inflammation, polyclonal lymphoproliferation, and a wide spectrum of symptoms caused by a cytokine storm often including IL-6 and vascular endothelial growth factor (VEGF). Clinical hallmarks include fever, night sweats, lymphadenopathy, ascites, hepatosplenomegaly, elevated C-reactive protein (CRP), hypoalbuminemia, and anemia. Some patients experience mild flulike symptoms, whereas others experience severe sepsislike multiple organ system failure, anasarca, and death.
The recently described “TAFRO syndrome” identifies a subset of iMCD patients with shared manifestations, including thrombocytope- nia, anasarca/ascites, reticulin fibrosis in bone marrow, renal dysfunction, organomegaly (TAFRO), and typically normal immuno- globulin levels. Although first described in Japan in 2010, iMCD patients with TAFRO features have been observed around the world for decades. iMCD patients without TAFRO syndrome typically have thrombocytosis, hypergammaglobulinemia, and less severe fluid accumulation. This non-TAFRO group has been called idiopathic plasmacytic lymphadenopathy with polyclonal hyperimmunoglobulinemia or IPL-type.
There are an estimated 6500 to 7700 new CD cases diagnosed/ year in the United States, with 1650 cases of MCD. iMCD accounts for 33% to 58% of published MCD cases. iMCD can occur in individuals of any age with a range of 2 to 80 years (median, 50). Historically, 35% die within 5 years of diagnosis, 60% die within 10 years, and patients have a threefold increased prevalence of malignancy. Corticosteroids, rituximab, cytotoxic chemotherapy, immunosuppressants, immunomodulators, and anti–IL-6 therapies have all been reported for the treatment of iMCD. Antibodies targeting IL-6 (siltuximab) or the IL-6 co- receptor, gp80 (tocilizumab), can reverse symptoms in many patients and may improve long-term outcomes. Siltuximab was recently approved for iMCD based on results from an international randomized, controlled trial in which 34% of patients attained a complete or partial response compared with 0% on placebo. However, the lack of defined diagnostic criteria or disease-specific biomarkers can impede timely administration of treatment before organ dysfunction and death may occur. Clinicopathologic diagnostic criteria are urgently needed to facilitate timely recognition, diagnostic workup, and research into pathogenesis and treatment. In this study, we present a multidisciplinary, evidence-based consensus diagnostic criteria for iMCD.
Research Methods:
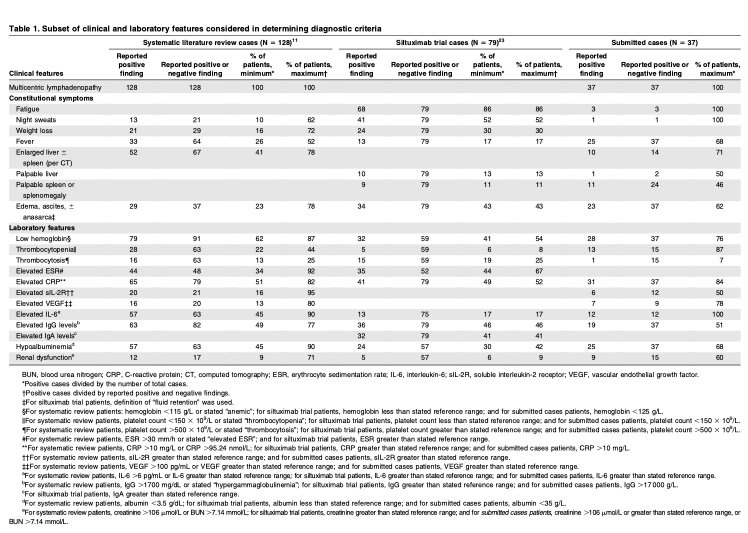
In 2013, the Castleman Disease Collaborative Network (CDCN)27 Scientific Advisory Board prioritized the establishment of an evidence-based, patient- guided, expert consensus diagnostic criteria. An international working group comprising 34 pediatric and adult hematopathology, hematology/oncology, rheumatology, immunology, and infectious diseases experts in iMCD and related disorders representing 8 countries on 5 continents, including 2 physicians that are also iMCD patients, was assembled (Figure 2). The CDCN assembled clinical data for 244 iMCD patients as well as 88 lymph node tissue biopsies for histopathologic review. One-hundred twenty-eight cases came from a systematic literature review of pathology-based iMCD, where HHV-8 was excluded and individual clinical data were available, 37 cases were submitted by working group members, and 79 were from a randomized controlled study of siltuximab in subjects with symptomatic iMCD (NCT01024036). Cases with ,80 k/mL platelets, elevated transaminases, and/or kidney dysfunction were excluded from NCT01024036, and (46/79) 58% of included cases received treatment before enrollment.
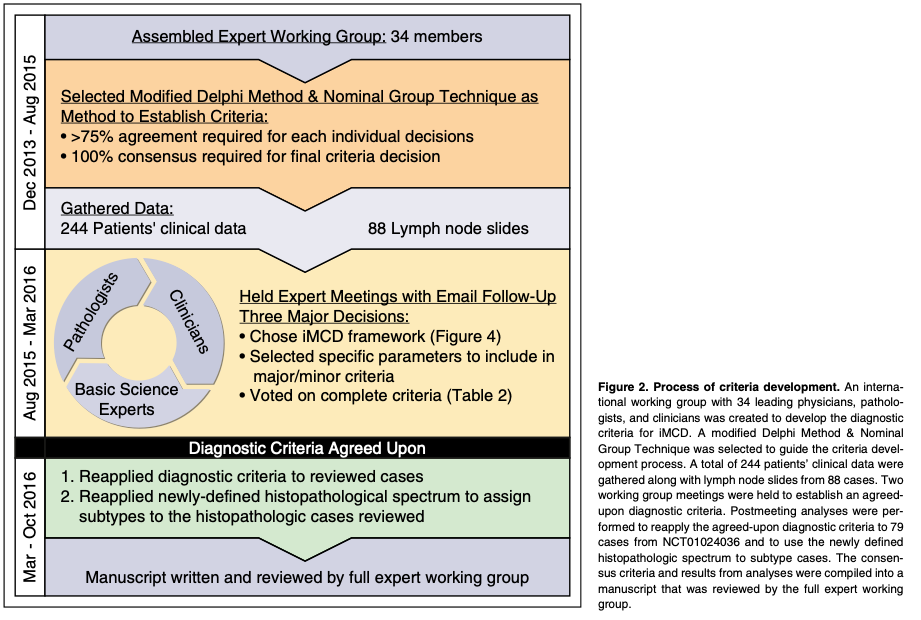
An international symposium sponsored by the CDCN and University of Pennsylvania Orphan Disease Center was held on November 20-21, 2015 in Philadelphia, Pennsylvania with 21 expert participants, and a follow-up meeting was held on December 6, 2015 in Orlando, Florida with 19 participants. All votes were anonymous and .75% agreement was needed to pass an individual decision. The final criteria vote required 100% consensus.
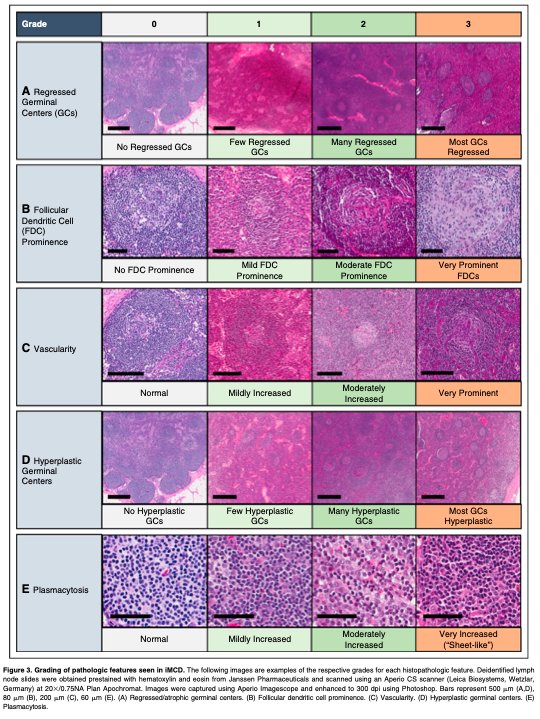
Literature reviews and expert interviews were performed to select a hybrid Delphi method and Nominal Group Technique (NGT) approach28 to guide criteria development. Clinical and laboratory parameters were chosen for consideration from literature review and expert nomination via the Delphi method in advance of the meetings (Table 1). NGT was used during the meetings to select parameters through group discussion and secret ballots and to achieve consensus. A team of expert hematopathologists examined hematoxylin and eosin–stained lymph node slides from 88 cases with a presumptive diagnosis of iMCD and graded the following histopathologic features using a scale of 0-3: regressed germinal centers (GCs), follicular dendritic cell (FDC) prominence, vascular proliferation, plasmacytosis, and hyperplastic GCs (Figure 3). The team expanded during the working group meeting to include additional hematopa- thologists. The group reviewed each case simultaneously at a multihead microscope until a majority of reviewers voted on a grade for each feature. The average grade for each histopathologic feature assigned during review was calculated and compared between subtypes by 2-way analysis of variance using a generalized linear model. Three of the 88 submitted pathology cases had insufficient tissue to be fully assessed.
At the conclusion of the meetings, the newly established diagnostic criteria were applied separately to cases that met both Major Criteria from the literature review, submitted cases, and NCT01024036, to evaluate the number of reported Minor Criteria required for the case definition. We also calculated response to siltuximab in NCT01024036 based on the number of Minor Criteria. We calculated summary statistics by tabulation and percentages. Fisher’s exact test was used to assess significant differences in treatment response rate. A value of P < .05 was considered significant. Statistical tests were performed using SAS 9.4.
Results and Discussion:
If you are interested in more information, see our paper here!