Projects
General
Many aspects of lipid metabolism in liver are controlled by the nuclear receptor PPARα, a transcription factor which functions by binding DNA and regulating gene expression. PPARα is essential for the response to fasting, in which the liver supplies energy to other tissues. In fasting, adipose tissue releases free fatty acids into the blood, and PPARα is the major activator of liver fatty acid uptake, transport, and β-oxidation, which provides the substrate for ketogenesis and energy for gluconeogenesis. Indeed, mice lacking PPARα respond poorly to fasting with markedly fatty livers but low glucose and ketones. PPARα also regulates genes involved in other lipid metabolic processes involving lipoproteins, lipid droplets, and bile acids. The effect on lipoproteins is exploited by the fibrate class of PPARα ligand drugs, which are used clinically in dyslipidemia to decrease serum triglycerides (TG, mainly in liver-derived VLDL particles) and raise HDL “good” cholesterol. PPARα is thus broadly relevant to cardiometabolic disease including hypertriglyceridemia and non-alcoholic fatty liver disease, and we hypothesize that regulatory variation in PPARα sites contributes to disease risk and treatment.
Close
Project 1: PPARα regulatory variation in fasting and fibrate drug response
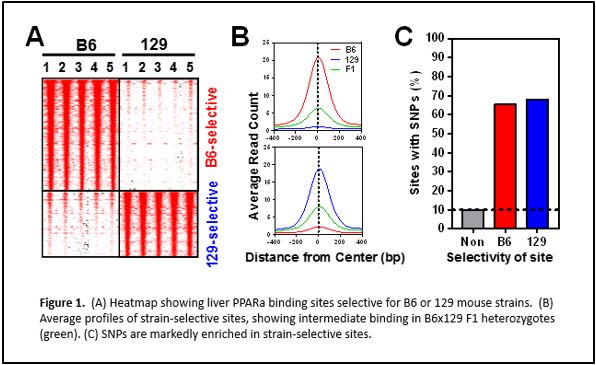
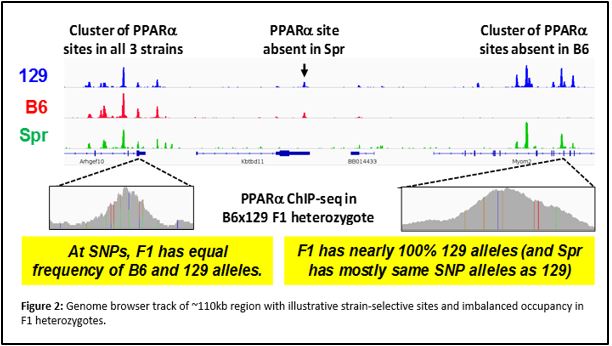
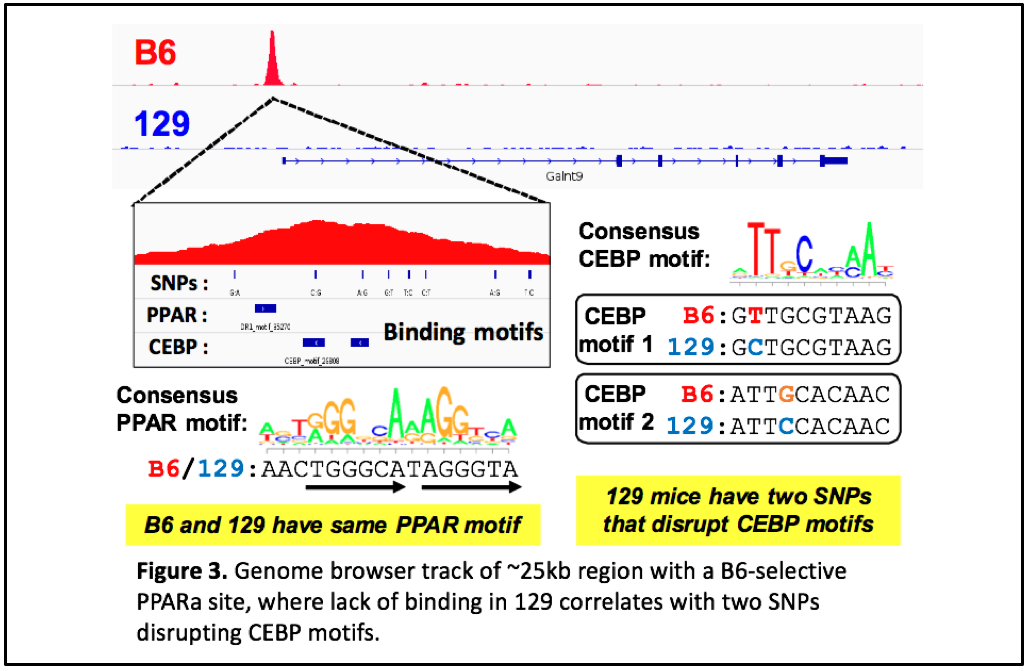
We have performed chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) to identify PPARα binding sites genome-wide in two mouse strains with different metabolic disease susceptibility: C57BL/6J (B6) and 129S1/SvImJ (129). We have also subjected these mice to conditions that activate PPARα physiologically (by fasting) or pharmacologically (by agonist drugs like fenofibrate), and surveyed differences in hepatic gene expression by RNA-seq. Integrative genomic analysis has revealed candidate genes that differ between the strains in their transcriptional regulation by PPARα. Ongoing studies will demonstrate the relevance of these variable PPARα regulatory sites to physiology and drug response.
Close
Project 2: PPARα regulatory variation in non-alcoholic fatty liver disease (NAFLD)
The worldwide epidemic of obesity has led to drastic increases in the prevalence of type 2 diabetes and other metabolic diseases, and non-alcoholic fatty liver disease (NAFLD) is a relatively underappreciated manifestation of obesity and insulin resistance. Simple liver steatosis may be nearly universal in obese individuals, and 10-20% progress to liver inflammation (non-alcoholic steatohepatitis, NASH). Some with NASH progress further to liver fibrosis and cirrhosis, which causes serious morbidity and mortality due to liver failure and hepatocellular carcinoma. From 2001-2009, the number of liver transplants due to NAFLD increased by 10-fold, and NAFLD may soon exceed viral hepatitis and even alcohol abuse as indications for liver transplant. There is an urgent need for better understanding of NAFLD, as there are no clinical predictors for which patients will progress to serious liver disease, and no FDA-approved treatments for NASH.
PPARα has been linked by others to NAFLD in the widely-used mouse model of feeding a methionine-choline deficient (MCD) diet. Mice lacking PPARα develop worse liver disease, and PPARα agonist drugs prevent or reverse liver damage. We aim to elucidate the mechanisms of PPARα benefits on NAFLD, and identify mouse strain effects due to natural regulatory variation. First, we are feeding MCD diet to B6 wild type mice, as well as PPARα null mice, then profiling PPARα genomic occupancy and gene regulation, to identify changes during MCD-induced liver damage. Second, we will compare the effects of MCD on 129 strain mice, to test whether PPARα regulatory variation underlies difference in liver disease development. Third, we will treat B6 and 129 mice on MCD diet with PPARα agonist drugs, to identify strain-specific responses to treatment.
Close
Project 3: PPARα regulatory variation in human dyslipidemia and fatty liver disease
We are obtaining human liver samples to identify PPARα binding events that differ among in individuals due to genetic variants such as single nucleotide polymorphisms (SNPs). Once we identify these common variants affecting PPARα binding, we will integrate them with published human genetic studies to test whether these SNPs cause differences in cardiometabolic phenotypes, in particular serum triglyceride levels and the response to fibrate drugs. Furthermore, we will obtain liver samples spanning the spectrum of NAFLD (healthy, steatotic, NASH, fibrosis/cirrhosis) to study the effects of disease progression on PPARα binding and gene regulation. Candidate disease-related genes and variants identified in these human studies will be studied in mouse models, or in human models including induced pluripotent cells and genome editing approaches.
Close
Contact Us
Soccio Lab
Smilow Center for Translational Research
3400 Civic Center Blvd
12th floor
Philadelphia, PA 19104
Office: room 12-112
Lab: room 12-178
215-573-4598
Email Us
