Publications
-
 Lineage-resolved analysis of embryonic gene expression evolution in C. elegans and C. briggsaeRead More about
Lineage-resolved analysis of embryonic gene expression evolution in C. elegans and C. briggsaeRead More about
Science, June 2025 *free access using this link*
Lineage-resolved analysis of embryonic gene expression evolution in C. elegans and C. briggsae
Science, June 2025 *free access using this link*
Large C, Khanal R, Hillier L, Huynh C, Kubo C, Kim J, Kubo C, Waterston R, Murray JI
Link to Full Article
Abstract
The constraints that govern the evolution of gene expression patterns across development remain unclear. Single-cell RNA sequencing can detail these constraints by systematically profiling homologous cells. The conserved invariant embryonic lineage of Caenorhabditis elegans and C. briggsae makes them ideal for comparing cell type gene expression across evolution. Measuring the spatiotemporal divergence of gene expression across embryogenesis, we find a high level of similarity in gene expression programs between species despite tens of millions of years of evolutionary divergence. Nonetheless, thousands of genes show divergence in their cell type specific expression patterns, with enrichment for functions in environmental response and behavior. Neuronal cell types show higher divergence than others such as the intestine and germline. This work identifies likely constraints on the evolution of developmental gene expression.
-
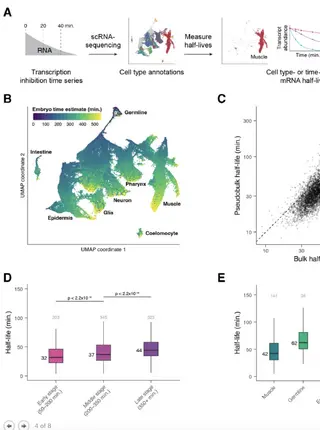 A spatiotemporally resolved atlas of mRNA decay in the C. elegans embryo reveals differential regulation of mRNA stability across stages and cell typesRead More about
A spatiotemporally resolved atlas of mRNA decay in the C. elegans embryo reveals differential regulation of mRNA stability across stages and cell typesRead More about
Genome Research, Jan 2024
A spatiotemporally resolved atlas of mRNA decay in the C. elegans embryo reveals differential regulation of mRNA stability across stages and cell types
Genome Research, Jan 2024
Peng F, Nordgren E, Murray JI
Link to Full Article
Abstract
During embryonic development, cells undergo dynamic changes in gene expression that are required for appropriate cell fate specification. Although both transcription and mRNA degradation contribute to gene expression dynamics, patterns of mRNA decay are less well-understood. Here we directly measured spatiotemporally resolved mRNA decay rates transcriptome-wide throughout C. elegans embryogenesis by transcription inhibition followed by bulk and single-cell RNA-sequencing. This allowed us to calculate mRNA half-lives within specific cell types and developmental stages and identify differentially regulated mRNA decay throughout embryonic development. We identified transcript features that are correlated with mRNA stability and found that mRNA decay rates are associated with distinct peaks in gene expression over time. Moreover, we provide evidence that, on average, mRNA is more stable in the germline compared to in the soma and in later embryonic stages compared to in earlier stages. This work suggests that differential mRNA decay across cell states and time helps to shape developmental gene expression, and it provides a valuable resource for studies of mRNA turnover regulatory mechanisms.
-
Read More about
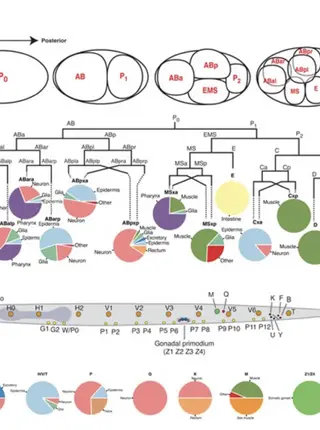 Mechanisms of lineage specification in Caenorhabditis elegans
Mechanisms of lineage specification in Caenorhabditis elegans
Genetics. Dec 2023
Liu J, Murray JI.
Link to Full Article
Abstract
The studies of cell fate and lineage specification are fundamental to our understanding of the development of multicellular organisms. Caenorhabditis elegans has been one of the premiere systems for studying cell fate specification mechanisms at single cell resolution, due to its transparent nature, the invariant cell lineage, and fixed number of somatic cells. We discuss the general themes and regulatory mechanisms that have emerged from these studies, with a focus on somatic lineages and cell fates. We next review the key factors and pathways that regulate the specification of discrete cells and lineages during embryogenesis and postembryonic development; we focus on transcription factors and include numerous lineage diagrams that depict the expression of key factors that specify embryonic founder cells and postembryonic blast cells, and the diverse somatic cell fates they generate. We end by discussing some future perspectives in cell and lineage specification.
-
Read More about
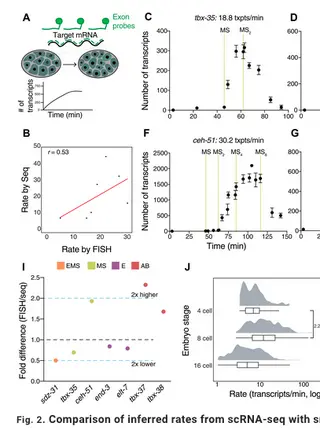 Transcript accumulation rates in the early Caenorhabditis elegans embryo
Transcript accumulation rates in the early Caenorhabditis elegans embryo
Science Advances, Aug 2023
Sivaramakrishnan P, Watkins C, Murray JI
Link to Full Article
Abstract
Dynamic transcriptional changes are widespread in rapidly dividing developing embryos when cell fate decisions are made quickly. The Caenorhabditis elegans embryo overcomes these constraints partly through the rapid production of high levels of transcription factor mRNAs. Transcript accumulation rates for some developmental genes are known at single-cell resolution, but genome-scale measurements are lacking. We estimate zygotic mRNA accumulation rates from single-cell RNA sequencing data calibrated with single-molecule transcript imaging. Rapid transcription is common in the early C. elegans embryo with rates highest soon after zygotic transcription begins. High-rate genes are enriched for recently duplicated cell-fate regulators and share common genomic features. We identify core promoter elements associated with high rate and measure their contributions for two early endomesodermal genes, ceh-51 and sdz-31. Individual motifs modestly affect accumulation rates, suggesting multifactorial control. These results are a step toward estimating absolute transcription kinetics and understanding how transcript dosage drives developmental decisions.
-
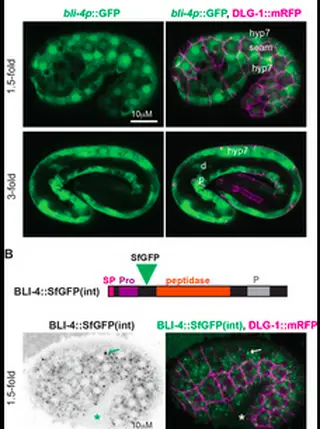 The proprotein convertase BLI-4 promotes collagen secretion during assembly of the Caenorhabditis elegans cuticleRead More about
The proprotein convertase BLI-4 promotes collagen secretion during assembly of the Caenorhabditis elegans cuticleRead More about
bioRxiv, June 2023
The proprotein convertase BLI-4 promotes collagen secretion during assembly of the Caenorhabditis elegans cuticle
bioRxiv, June 2023
Birnbaum SK, Cohen JD, Belfi A, Murray JI, Adams JRG, Chisholm AD, Sundaram MV
Link to Full Article
Abstract
Some types of collagens, including transmembrane MACIT collagens and C. elegans cuticle collagens, are N-terminally cleaved at a dibasic site that resembles the consensus for furin or other proprotein convertases of the subtilisin/kexin (PCSK) family. Such cleavage may release transmembrane collagens from the plasma membrane and affect extracellular matrix assembly or structure. However, the functional consequences of such cleavage are unclear and evidence for the role of specific PCSKs is lacking. Here, we used endogenous collagen fusions to fluorescent proteins to visualize the secretion and assembly of the first collagen-based cuticle in C. elegans and then tested the role of the PCSK BLI-4 in these processes. Unexpectedly, we found that cuticle collagens SQT-3 and DPY-17 are secreted into the extra embryonic space several hours before cuticle matrix assembly. Furthermore, this early secretion depends on BLI-4/PCSK; in bli-4 and cleavage-site mutants, SQT-3 and DPY-17 are not efficiently secreted and instead form large intracellular aggregates. Their later assembly into cuticle matrix is reduced but not entirely blocked. These data reveal a role for collagen N-terminal processing in intracellular trafficking and in the spatial and temporal restriction of matrix assembly in vivo . Our observations also prompt a revision of the classic model for C. elegans cuticle matrix assembly and the pre-cuticle-to-cuticle transition, suggesting that cuticle layer assembly proceeds via a series of regulated steps and not simply by sequential secretion and deposition.
-
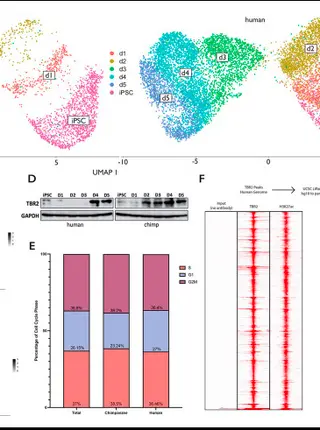 Young transposable elements rewired gene regulatory networks in human and chimpanzee hippocampal intermediate progenitorsRead More about
Young transposable elements rewired gene regulatory networks in human and chimpanzee hippocampal intermediate progenitorsRead More about
Development, Oct 2022
Young transposable elements rewired gene regulatory networks in human and chimpanzee hippocampal intermediate progenitors
Development, Oct 2022
Patoori S, Barnada SM, Large C, Murray JI, Trizzino M
Link to Full Article
Abstract
The hippocampus is associated with essential brain functions, such as learning and memory. Human hippocampal volume is significantly greater than expected compared with that of non-human apes, suggesting a recent expansion. Intermediate progenitors, which are able to undergo multiple rounds of proliferative division before a final neurogenic division, may have played a role in evolutionary hippocampal expansion. To investigate the evolution of gene regulatory networks underpinning hippocampal neurogenesis in apes, we leveraged the differentiation of human and chimpanzee induced pluripotent stem cells into TBR2 (or EOMES)-positive hippocampal intermediate progenitor cells (hpIPCs). We found that the gene networks active in hpIPCs are significantly different between humans and chimpanzees, with ∼2500 genes being differentially expressed. We demonstrate that species-specific transposon-derived enhancers contribute to these transcriptomic differences. Young transposons, predominantly endogenous retroviruses and SINE-Vntr-Alus (SVAs), were co-opted as enhancers in a species-specific manner. Human-specific SVAs provided substrates for thousands of novel TBR2-binding sites, and CRISPR-mediated repression of these SVAs attenuated the expression of ∼25% of the genes that are upregulated in human intermediate progenitors relative to the same cell population in the chimpanzee.
-
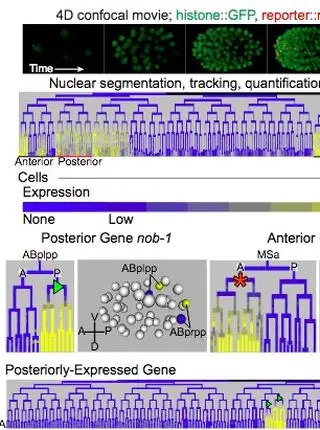 pop-1/TCF, ref-2/ZIC and T-box factors regulate the development of anterior cells in the C. elegans embryoRead More about
pop-1/TCF, ref-2/ZIC and T-box factors regulate the development of anterior cells in the C. elegans embryoRead More about
Dev Biol, Sept 2022
pop-1/TCF, ref-2/ZIC and T-box factors regulate the development of anterior cells in the C. elegans embryo
Dev Biol, Sept 2022
Rumley J, Preston E, Cook D, Peng F, Zacharias A, Wu L, Jileaeva I, Murray JI
Link to Full Article
Abstract
Patterning of the anterior-posterior axis is fundamental to animal development. The Wnt pathway plays a major role in this process by activating the expression of posterior genes in animals from worms to humans. This observation raises the question of whether the Wnt pathway or other regulators control the expression of the many anterior-expressed genes. We found that the expression of five anterior-specific genes in Caenorhabditis elegans embryos depends on the Wnt pathway effectors pop-1/TCF and sys-1/β-catenin. We focused further on one of these anterior genes, ref-2/ZIC, a conserved transcription factor expressed in multiple anterior lineages. Live imaging of ref-2 mutant embryos identified defects in cell division timing and position in anterior lineages. Cis-regulatory dissection identified three ref-2 transcriptional enhancers, one of which is necessary and sufficient for anterior-specific expression. This enhancer is activated by the T-box transcription factors TBX-37 and TBX-38, and surprisingly, concatemerized TBX-37/38 binding sites are sufficient to drive anterior-biased expression alone, despite the broad expression of TBX-37 and TBX-38. Taken together, our results highlight the diverse mechanisms used to regulate anterior expression patterns in the embryo.
-
Read More about
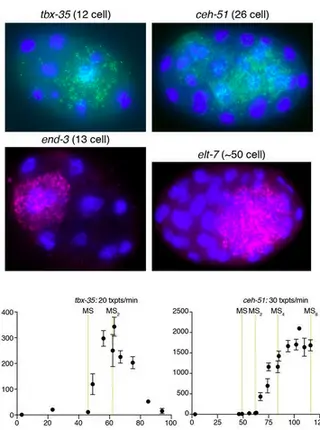 Transcript accumulation rates in the early C. elegans embryo
Transcript accumulation rates in the early C. elegans embryo
Sci Adv, Aug 2023
Sivaramakrishnan P, Watkins C, Murray JI
Link to Full Article
Abstract
Dynamic changes in transcription are widespread in the developing embryo, where cell cycles are rapid and cell fate decisions sometimes need to be made quickly, before the next cell division. In the early Caenorhabditis elegans embryo, specification of the intestine relies on high absolute levels of transcription factors that are a part of the gut gene regulatory network. These absolute levels are likely achieved by controlled transcript accumulation rates. However, accumulation rates have not been measured globally in the worm embryo. We used single cell RNA-seq data from the early C. elegans embryo to estimate the accumulation rates of zygotic genes up to the 24-cell stage. We find that rapid transcript accumulation is a characteristic feature of transcription factors across different cell types and lineages. We identified genomic features associated with high transcription rates and core promoter motifs that might drive these rates. For one Very High-rate gene ceh-51, which is required for mesoderm development, we measured the contributions of core promoter elements to rate. We find that each of these motifs contribute modestly to the accumulation rate of ceh-51, suggesting a complex relationship between promoter motifs and gene structure in controlling transcript accumulation rates. These results are a step towards understanding the regulation of transcript accumulation rates during embryonic cell fate specification.
-
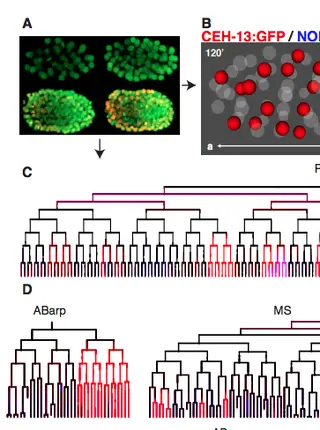 The anterior Hox gene ceh-13 and elt-1/GATA activate the posterior Hox genes nob-1 and php-3 to specify posterior lineages in the C. elegans embryoRead More about
The anterior Hox gene ceh-13 and elt-1/GATA activate the posterior Hox genes nob-1 and php-3 to specify posterior lineages in the C. elegans embryoRead More about
PLoS Genetics, May 2022
The anterior Hox gene ceh-13 and elt-1/GATA activate the posterior Hox genes nob-1 and php-3 to specify posterior lineages in the C. elegans embryo
PLoS Genetics, May 2022
Murray JI, Preston E, Crawford J, Rumley J, Amom P, Anderson B, Sivaramakrishnan P, Patel S, Bennett B, Lavon T, Peng F, Zacharias A
Link to Full Article
Abstract
Hox transcription factors play a conserved role in specifying positional identity during animal development, with posterior Hox genes typically repressing the expression of more anterior Hox genes. Here, we dissect the regulation of the posterior Hox genes nob-1 and php-3 in the nematode C. elegans. We show that nob-1 and php-3 are co-expressed in gastrulation-stage embryos in cells that express the anterior Hox gene ceh-13. This expression is controlled by several partially redundant transcriptional enhancers. Surprisingly, these enhancers require ceh-13 for expression, providing an example of an anterior Hox gene positively regulating a posterior Hox gene. Several other regulators also act positively through nob-1/php-3 enhancers, including elt-1/GATA, ceh-20/ceh-40/Pbx, unc-62/Meis, pop-1/TCF, ceh-36/Otx and unc-30/Pitx. We identified defects in both cell position and cell division patterns in ceh-13 and nob-1;php-3 mutants, suggesting that these factors regulate lineage identity in addition to positional identity. Together, our results highlight the complexity and remarkable flexibility of Hox gene regulation and function.
-
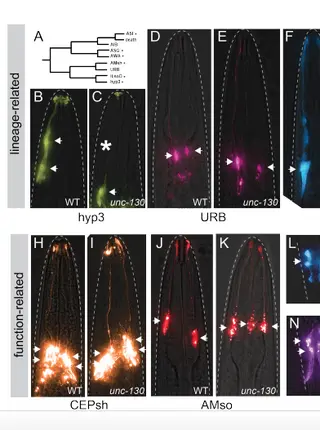 Lineage-specific control of convergent differentiation by a Forkhead repressorRead More about
Lineage-specific control of convergent differentiation by a Forkhead repressorRead More about
Development, Oct 2021
Lineage-specific control of convergent differentiation by a Forkhead repressor
Development, Oct 2021
Mizeracka K, Rogers J, Rumley J, Shaham S, Bulyk M, Murray JI, Heiman M
Link to Full Article
Abstract
During convergent differentiation, multiple developmental lineages produce a highly similar or identical cell type. However, the molecular players that drive convergent differentiation are not known. Here, we show that the C. elegans Forkhead transcription factor UNC-130 is required in only one of three convergent lineages that produce the same glial cell type. UNC-130 acts transiently as a repressor in progenitors and newly-born terminal cells to allow the proper specification of cells related by lineage rather than by cell type. Specification defects correlate with UNC-130:DNA binding, and UNC-130 can be functionally replaced by its human homolog, the neural crest lineage determinant FoxD3. We propose that, in contrast to terminal selectors that activate cell-type specific transcriptional programs in terminally differentiating cells, UNC-130 acts earlier to enable molecularly distinct progenitors to produce equivalent cell types. These findings provide evidence that convergent differentiation involves distinct transcriptional paths leading to the same cell type.
-
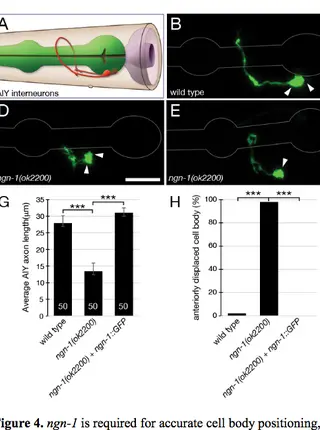 ngn-1/neurogenin activates transcription of multiple terminal selector transcription factors in the Caenorhabditis elegans nervous system.Read More about
ngn-1/neurogenin activates transcription of multiple terminal selector transcription factors in the Caenorhabditis elegans nervous system.Read More about
G3: Genes, Genomics, Genetics. April 2020.
ngn-1/neurogenin activates transcription of multiple terminal selector transcription factors in the Caenorhabditis elegans nervous system.
G3: Genes, Genomics, Genetics. April 2020.
Christensen EL, Beasley A, Radchuk J, Mielko ZE, Preston E, Stuckett E, Murray JI, Hudson MLink to Full Article
Abstract
Proper nervous system development is required for an organism's survival and function. Defects in neurogenesis have been linked to neurodevelopmental disorders such as schizophrenia and autism. Understanding the gene regulatory networks that orchestrate neural development, specifically cascades of proneural transcription factors, can better elucidate which genes are most important during early neurogenesis. Neurogenins are a family of deeply conserved factors shown to be both necessary and sufficient for the development of neural subtypes. However, the immediate downstream targets of neurogenin are not well characterized. The objective of this study was to further elucidate the role of ngn-1/neurogenin in nervous system development and to identify its downstream transcriptional targets, using the nematode Caenorhabditis elegans as a model for this work. We found that ngn-1 is required for axon outgrowth, nerve ring architecture, and neuronal cell fate specification. We also showed that ngn-1 may have roles in neuroblast migration and epithelial integrity during embryonic development. Using RNA sequencing and comparative transcriptome analysis, we identified eight transcription factors (hlh-34/NPAS1, unc-42/PROP1, ceh-17/PHOX2A, lim-4/LHX6, fax-1/NR2E3, lin-11/LHX1, tlp-1/ZNF503, and nhr-23/RORB) whose transcription is activated, either directly or indirectly, by ngn-1. Our results show that ngn-1 has a role in transcribing known terminal regulators that establish and maintain cell fate of differentiated neural subtypes and confirms that ngn-1 functions as a proneural transcription factor in C. elegans neurogenesis.
-
Read More about
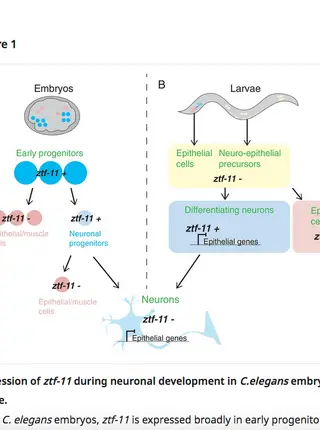 Neurogenesis: Silencing the alternative
Neurogenesis: Silencing the alternative
eLife. Aug 2019.
Sivaramakrishnan, P, Murray, JI.
Link to Full Article
Abstract
The transcription factor ztf-11 promotes neuronal differentiation by repressing other cell fates in the nematode worm C. elegans.
-
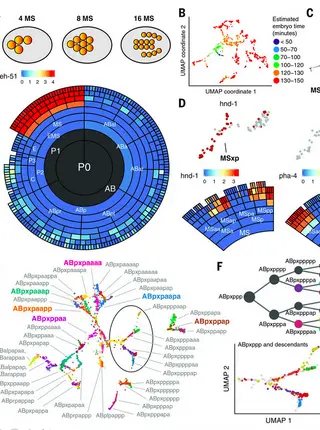 A lineage-resolved molecular atlas of C. elegans embryogenesis at single-cell resolutionRead More about
A lineage-resolved molecular atlas of C. elegans embryogenesis at single-cell resolutionRead More about
Science. Sep 2019.
A lineage-resolved molecular atlas of C. elegans embryogenesis at single-cell resolution
Science. Sep 2019.
Packer JS, Zhu Q, Huynh C, Sivaramakrishnan P, Preston E, Dueck H, Stefanik D, Tan K, Trapnell C, Kim J, Waterston R, Murray JI.
Link to Full Article
Abstract
Caenorhabditis elegans is an animal with few cells but a wide diversity of cell types. In this study, we characterize the molecular basis for their specification by profiling the transcriptomes of 86,024 single embryonic cells. We identify 502 terminal and preterminal cell types, mapping most single-cell transcriptomes to their exact position in C. elegans’ invariant lineage. Using these annotations, we find that (i) the correlation between a cell’s lineage and its transcriptome increases from middle to late gastrulation, then falls substantially as cells in the nervous system and pharynx adopt their terminal fates; (ii) multilineage priming contributes to the differentiation of sister cells at dozens of lineage branches; and (iii) most distinct lineages that produce the same anatomical cell type converge to a homogenous transcriptomic state.
-
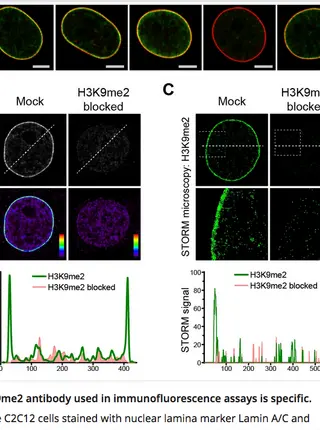 H3K9me2 orchestrates inheritance of spatial positioning of peripheral heterochromatin through mitosis.Read More about
H3K9me2 orchestrates inheritance of spatial positioning of peripheral heterochromatin through mitosis.Read More about
eLife. June 2019.
H3K9me2 orchestrates inheritance of spatial positioning of peripheral heterochromatin through mitosis.
eLife. June 2019.
Poleshko, A, Smith, CL, Nguyen, SC, Sivaramakrishnan, P, Murray, JI, Lakadamyali, M, Joyce, EF, Jain, R, Epstein, J.
Link to Full Article
Abstract
Cell-type-specific 3D organization of the genome is unrecognizable during mitosis. It remains unclear how essential positional information is transmitted through cell division such that a daughter cell recapitulates the spatial genome organization of the parent. Lamina-associated domains (LADs) are regions of repressive heterochromatin positioned at the nuclear periphery that vary by cell type and contribute to cell-specific gene expression and identity. Here we show that histone 3 lysine 9 dimethylation (H3K9me2) is an evolutionarily conserved, specific mark of nuclear peripheral heterochromatin and that it is retained through mitosis. During mitosis, phosphorylation of histone 3 serine 10 temporarily shields the H3K9me2 mark allowing for dissociation of chromatin from the nuclear lamina. Using high-resolution 3D immuno-oligoFISH, we demonstrate that H3K9me2-enriched genomic regions, which are positioned at the nuclear lamina in interphase cells prior to mitosis, re-associate with the forming nuclear lamina before mitotic exit. The H3K9me2 modification of peripheral heterochromatin ensures that positional information is safeguarded through cell division such that individual LADs are re-established at the nuclear periphery in daughter nuclei. Thus, H3K9me2 acts as a 3D architectural mitotic guidepost. Our data establish a mechanism for epigenetic memory and inheritance of spatial organization of the genome.
-
Read More about
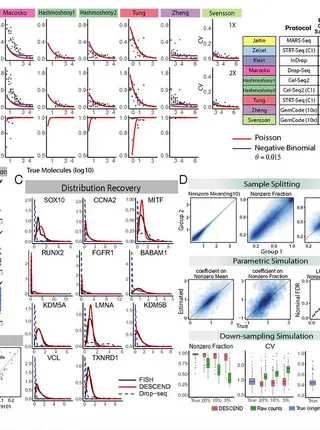 Gene expression distribution deconvolution in single-cell RNA sequencing.
Gene expression distribution deconvolution in single-cell RNA sequencing.
PNAS. July 2018
Wang, J, Huang, M, Torre, E, Dueck, H, Shaffer, S, Murray, J, Raj, A, Li, M, Zhang, NR.
Link to Full Article
Abstract
Single-cell RNA sequencing (scRNA-seq) enables the quantification of each gene’s expression distribution across cells, thus allowing the assessment of the dispersion, nonzero fraction, and other aspects of its distribution beyond the mean. These statistical characterizations of the gene expression distribution are critical for understanding expression variation and for selecting marker genes for population heterogeneity. However, scRNA-seq data are noisy, with each cell typically sequenced at low coverage, thus making it difficult to infer properties of the gene expression distribution from raw counts. Based on a reexamination of nine public datasets, we propose a simple technical noise model for scRNA-seq data with unique molecular identifiers (UMI). We develop deconvolution of single-cell expression distribution (DESCEND), a method that deconvolves the true cross-cell gene expression distribution from observed scRNA-seq counts, leading to improved estimates of properties of the distribution such as dispersion and nonzero fraction. DESCEND can adjust for cell-level covariates such as cell size, cell cycle, and batch effects. DESCEND’s noise model and estimation accuracy are further evaluated through comparisons to RNA FISH data, through data splitting and simulations and through its effectiveness in removing known batch effects. We demonstrate how DESCEND can clarify and improve downstream analyses such as finding differentially expressed genes, identifying cell types, and selecting differentiation markers.
-
Read More about
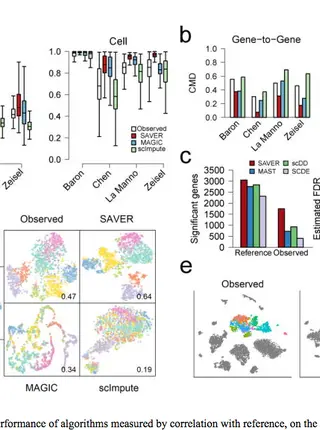 SAVER: gene expression recovery for single-cell RNA sequencing.
SAVER: gene expression recovery for single-cell RNA sequencing.
Nature Methods. July 2018.
Huang, M, Wang, J, Torre, E, Dueck, H, Shaffer, S, Bonasio, R, Murray, J, Raj, A, Li, M, Zhang, N.R
Link to Full Article
Abstract
In single-cell RNA sequencing (scRNA-seq) studies, only a small fraction of the transcripts present in each cell are sequenced. This leads to unreliable quantification of genes with low or moderate expression, which hinders downstream analysis. To address this challenge, we developed SAVER (single-cell analysis via expression recovery), an expression recovery method for unique molecule index (UMI)-based scRNA-seq data that borrows information across genes and cells to provide accurate expression estimates for all genes.
-
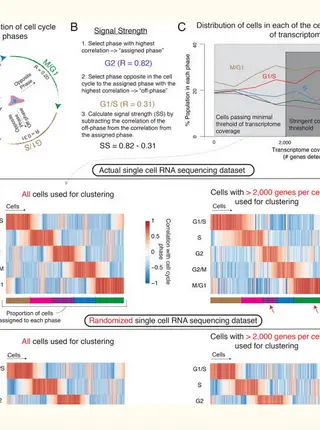 Rare Cell Detection by Single-Cell RNA Sequencing as Guided by Single-Molecule RNA FISH.Read More about
Rare Cell Detection by Single-Cell RNA Sequencing as Guided by Single-Molecule RNA FISH.Read More about
Cell Systems. Feb 2018.
Rare Cell Detection by Single-Cell RNA Sequencing as Guided by Single-Molecule RNA FISH.
Cell Systems. Feb 2018.
Torre E, Dueck H, Shaffer S, Gospocic J, Gupte R, Bonasio R, Kim J, Murray J, Raj A.
Link to Full Article
Abstract
Although single cell RNA sequencing can reliably detect large-scale transcriptional programs, it is unclear whether it accurately captures the behavior of individual genes, especially those that express only in rare cells. Here, we use single molecule RNA FISH as a gold standard to assess tradeoffs in single cell RNA sequencing data for detecting rare cell expression variability. We quantified the gene expression distribution for 26 genes that range from ubiquitous to rarely expressed and found that the correspondence between estimates across platforms improved with both transcriptome coverage and increased number of cells analyzed. Further, by characterizing the tradeoff between transcriptome coverage and number of cells analyzed, we show that when the number of genes required to answer a given biological question is small, then greater transcriptome coverage is more important than analyzing large numbers of cells. More generally, our report provides guidelines for selecting quality thresholds for single cell RNA sequencing experiments aimed at rare cell analyses.
-
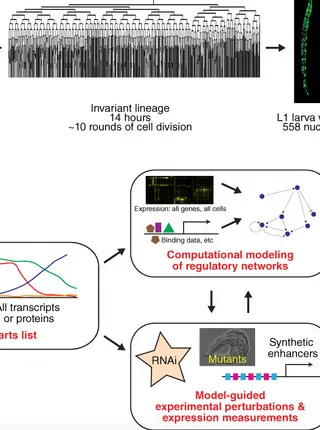 Systems biology of embryonic development: Prospects for a complete understanding of the Caenorhabditis elegans embryo.Read More about
Systems biology of embryonic development: Prospects for a complete understanding of the Caenorhabditis elegans embryo.Read More about
Wiley Online Library. Jan 2018.
Systems biology of embryonic development: Prospects for a complete understanding of the Caenorhabditis elegans embryo.
Wiley Online Library. Jan 2018.
Murray JI.
Link to Full Article
Abstract
The convergence of developmental biology and modern genomics tools brings the potential for a comprehensive understanding of developmental systems. This is especially true for the Caenorhabditis elegans embryo because its small size, invariant developmental lineage, and powerful genetic and genomic tools provide the prospect of a cellular resolution understanding of messenger RNA (mRNA) expression and regulation across the organism. We describe here how a systems biology framework might allow large‐scale determination of the embryonic regulatory relationships encoded in the C. elegans genome. This framework consists of two broad steps: (a) defining the “parts list”—all genes expressed in all cells at each time during development and (b) iterative steps of computational modeling and refinement of these models by experimental perturbation. Substantial progress has been made towards defining the parts list through imaging methods such as large‐scale green fluorescent protein (GFP) reporter analysis. Imaging results are now being augmented by high‐resolution transcriptome methods such as single‐cell RNA sequencing, and it is likely the complete expression patterns of all genes across the embryo will be known within the next few years. In contrast, the modeling and perturbation experiments performed so far have focused largely on individual cell types or genes, and improved methods will be needed to expand them to the full genome and organism. This emerging comprehensive map of embryonic expression and regulatory function will provide a powerful resource for developmental biologists, and would also allow scientists to ask questions not accessible without a comprehensive picture.
-
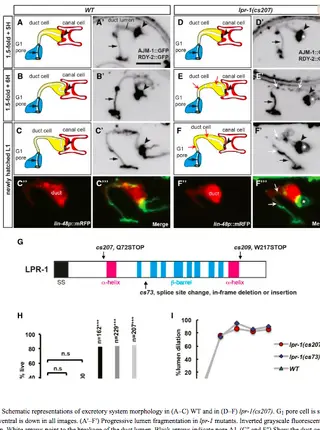 The Lipocalin LPR-1 Cooperates with LIN-3/EGF Signaling To Maintain Narrow Tube Integrity in Caenorhabditis elegans.Read More about
The Lipocalin LPR-1 Cooperates with LIN-3/EGF Signaling To Maintain Narrow Tube Integrity in Caenorhabditis elegans.Read More about
Genetics. March 2017.
The Lipocalin LPR-1 Cooperates with LIN-3/EGF Signaling To Maintain Narrow Tube Integrity in Caenorhabditis elegans.
Genetics. March 2017.
Pu, P, Stone, CE, Burdick, JT, Murray, JI, Sundaram, M.
Link to Full Article
Abstract
Lipocalins are secreted cup-shaped glycoproteins that bind sterols, fatty acids, and other lipophilic molecules. Lipocalins have been implicated in a wide array of processes related to lipophilic cargo transport, sequestration, and signaling, and several are used as biomarkers for human disease, but the functions of most lipocalins remain poorly understood. Here we show that the Caenorhabditis elegans lipocalin LPR-1 is required to maintain apical membrane integrity and a continuous lumen in two narrow unicellular tubes, the excretory duct and pore, during a period of rapid lumen elongation. LPR-1 fusion protein is expressed by the duct and pore and accumulates both intracellularly and in apical extracellular compartments, but it can also function cell nonautonomously when provided from outside of the excretory system. lpr-1 mutant defects can be rescued by increased signaling through the epidermal growth factor (EGF)-Ras-extracellular signal regulated kinase (ERK) pathway, which promotes the more elongated duct vs. less elongated pore tube fate. Spatial and temporal rescue experiments indicate that Ras signaling acts within the duct and pore tubes during or prior to cell fate determination to bypass the requirement for LPR-1 lpr-1 mutations did not disrupt LIN-3/EGF-dependent duct-fate specification, prevent functioning of any specific LIN-3/EGF isoform, or alter LET-23/EGFR localization, and reduced signaling did not phenocopy or enhance lpr-1 mutant defects. These data suggest that LPR-1 protects lumen integrity through a LIN-3/EGF-independent mechanism, but that increased signaling upregulates some target(s) that can compensate for lpr-1 absence.
-
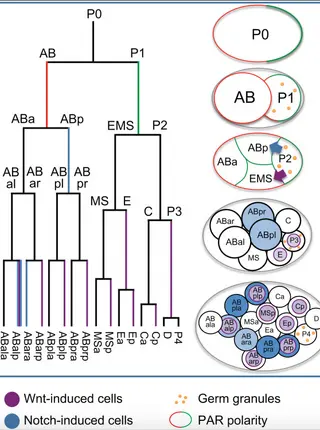 Combinatorial decoding of the invariant C. elegans embryonic lineage in space and time.Read More about
Combinatorial decoding of the invariant C. elegans embryonic lineage in space and time.Read More about
Genesis. Apr 2016.
Combinatorial decoding of the invariant C. elegans embryonic lineage in space and time.
Genesis. Apr 2016.
Zacharias AL, Murray JI.
Link to Full Article
Abstract
Understanding how a single cell, the zygote, can divide and differentiate to produce the diverse animal cell types is a central goal of developmental biology research. The model organism Caenorhabditis elegans provides a system that enables a truly comprehensive understanding of this process across all cells. Its invariant cell lineage makes it possible to identify all of the cells in each individual and compare them across organisms. Recently developed methods automate the process of cell identification, allowing high-throughput gene expression characterization and phenotyping at single cell resolution. In this Review, we summarize the sequences of events that pattern the lineage including establishment of founder cell identity, the signaling pathways that diversify embryonic fate, and the regulators involved in patterning within these founder lineages before cells adopt their terminal fates. We focus on insights that have emerged from automated approaches to lineage tracking, including insights into mechanisms of robustness, context-specific regulation of gene expression, and temporal coordination of differentiation. We suggest a model by which lineage history produces a combinatorial code of transcription factors that act, often redundantly, to ensure terminal fate.
-
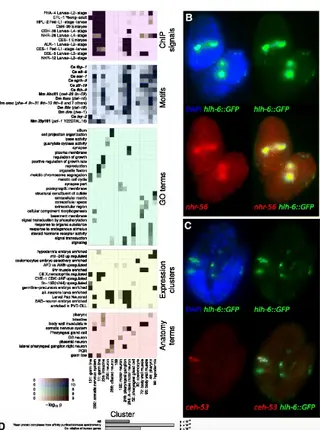 Overlapping cell population expression profiling and regulatory inference in C. elegans.Read More about
Overlapping cell population expression profiling and regulatory inference in C. elegans.Read More about
BMC Genomics. Feb 2016.
Overlapping cell population expression profiling and regulatory inference in C. elegans.
BMC Genomics. Feb 2016.
Burdick, JT, Walton, S, Preston, E, Zacharias, A, Raj, A, Murray JI
Link to Full Article
Abstract
BACKGROUND:
Understanding gene expression across the diverse metazoan cell types during development is critical to understanding their function and regulation. However, most cell types have not been assayed for expression genome-wide.
RESULTS:
We applied a novel approach we term "Profiling of Overlapping Populations of cells (POP-Seq)" to assay differential expression across all embryonic cells in the nematode Caenorhabditis elegans. In this approach, we use RNA-seq to define the transcriptome of diverse partially overlapping FACS-sorted cell populations. This identified thousands of transcripts differentially expressed across embryonic cells. Hierarchical clustering analysis identified over 100 sets of coexpressed genes corresponding to distinct patterns of cell type specific expression. We identified thousands of candidate regulators of these clusters based on enrichment of transcription factor motifs and experimentally determined binding sites.
-
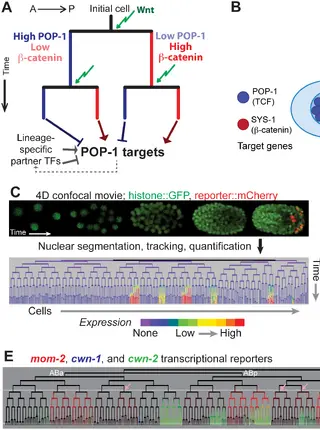 Quantitative Differences in Nuclear β-catenin and TCF Pattern Embryonic Cells in C. elegans.Read More about
Quantitative Differences in Nuclear β-catenin and TCF Pattern Embryonic Cells in C. elegans.Read More about
PLOS Genetics. Oct 2015.
Quantitative Differences in Nuclear β-catenin and TCF Pattern Embryonic Cells in C. elegans.
PLOS Genetics. Oct 2015.
Zacharias AL, Walton T, Preston E, Murray JI.
Link to Full Article
Abstract
The Wnt signaling pathway plays a conserved role during animal development in transcriptional regulation of distinct targets in different developmental contexts but it remains unclear whether quantitative differences in the nuclear localization of effector proteins TCF and β-catenin contribute to context-specific regulation. We investigated this question in Caenorhabditis elegans embryos by quantifying nuclear localization of fluorescently tagged SYS-1/β-catenin and POP-1/TCF and expression of Wnt ligands at cellular resolution by time-lapse microscopy and automated lineage tracing. We identified reproducible, quantitative differences that generate a subset of Wnt-signaled cells with a significantly higher nuclear concentration of the TCF/β-catenin activating complex. Specifically, β-catenin and TCF are preferentially enriched in nuclei of daughter cells whose parents also had high nuclear levels of that protein, a pattern that could influence developmental gene expression. Consistent with this, we found that expression of synthetic reporters of POP-1-dependent activation is biased towards cells that had high nuclear SYS-1 in consecutive divisions. We identified new genes whose embryonic expression patterns depend on pop-1. Most of these require POP-1 for either transcriptional activation or repression, and targets requiring POP-1 for activation are more likely to be expressed in the cells with high nuclear SYS-1 in consecutive divisions than those requiring POP-1 for repression. Taken together, these results indicate that SYS-1 and POP-1 levels are influenced by the parent cell’s SYS-1/POP-1 levels and this may provide an additional mechanism by which POP-1 regulates distinct targets in different developmental contexts.





















More Publications
(Murray lab members are in bold)
- Walton T, Preston E, Nair G, Zacharias AL, Raj A, Murray JI. The Bicoid Class Homeodomain Factors ceh-36/OTX and unc-30/PITX Cooperate in C. elegans Embryonic Progenitor Cells to Regulate Robust Development. PLoS Genetics. Mar 2015.
- Araya CL, Kawli T, Kundaje A, Jiang L, Wu B, Vafeados D, Terrell R, Weissdepp P, Gevirtzman L, Mace D, Niu W, Boyle AP, Xie D, Ma L, Murray JI, Reinke V, Waterston RH, Snyder M. Regulatory analysis of the C. elegans genome with spatiotemporal resolution. Nature. 2014.
- Churgin MA, He L, Murray JI, Fang-Yen C. Construction of a system for single-cell transgene induction in Caenorhabditis elegans using a pulsed infrared laser. Methods. 2014.
- Nair G, Walton T, Murray JI, Raj A. Gene transcription is coordinated with, but not dependent on, cell divisions during C. elegans embryonic fate specification. Development. 2013.
- Burdick JT, Murray JI. Deconvolution of gene expression from cell populations across the C. elegans lineage. BMC Bioinformatics. 2013.
- Richards J.L., Zacharias A.L., Walton T., Burdick J.T., Murray J.I. A quantitative model of normal Caenorhabditis elegans embryogenesis and its disruption after stress. Developmental Biology. 2013.
- SUPPLEMENTAL DATA: AceTreeModel.zip, Trees.zip
- Sarov M., Murray J.I., Schanze K., Pozniakovski A., Niu W., Angermann K., Hasse S., Rupprecht M., Vinis E., Tinney M., Preston E., Zinke A., Enst S., Teichgraber T., Janette J., Reis K., Janosch S., Schloissnig S., Ejsmont R.K., Slightam C., Xu X., Kim S.K., Reinke V., Stewart A.F., Snyder M., Waterston R.H., Hyman A.A. A genome-scale resource for in vivo tag-based protein function exploration in C. elegans. Cell. 2012.
- Murray J.I., Bao Z. Automated Lineage and Expression Profiling in Live Caenorhabditis elegans Embryos. Cold Spring Harbor Protocols. 2012.
- Li F., Zheng Q., Ryvkin P., Dragomir I., Desai Y., Aiyer S., Valladares O., Yang J., Bambina S., Sabin L.R., Murray J.I., Lamitina T., Raj A., Cherry S., Wang L.S., Gregory B.D. Global analysis of RNA secondary structure in two metazoans. Cell Reports. 2012.
- Abdus-Saboor I., Stone C.E., Murray J.I., Sundaram M.V. The Nkx5/HMX homeodomain protein MLS-2 is required for proper tube cell shape in the C. elegans excretory system. Developmental Biology. 2012.
- Murray J.I., Boyle T.J., Preston E., Vafeados D., Mericle B., Weisdepp P., Zhao Z., Bao Z., Boeck M., Waterston R.H. Multidimensional regulation of gene expression in the C. elegans embryo. Genome Research. 2012.
- Bao Z., Murray J.I. Mounting Caenorhabditis elegans embryos for live imaging of embryogenesis. Cold Spring Harbor Protocols. 2011.
- Boeck M.E., Boyle T., Bao Z., Murray J., Mericle B., Waterston R. Specific roles for the GATA transcription factors end-1 and end-3 during C. elegans E-lineage development. Developmental Biology. 2011.
- Abdus-Saboor I., Mancuso V.P., Murray J.I., Palozola K., Norris C., Hall D.H., Howell K., Huang K., Sundaram M.V. Notch and Ras promote sequential steps of excretory tube development in C. elegans. Development. 2011.
- Waterston et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010.
- Niu W., Lu Z..J, Zhong M., Sarov M., Murray J.I., Brdlik C.M., Janette J., Chen C., Alves P., Preston E., Slightham C., Jiang L., Hyman A.A., Kim S.K., Waterston R.H., Gerstein M., Snyder M., Reinke V. Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Research. 2011.
- Zhao, Z., Boyle, T., Murray, J.I., Wood, W., Waterston, RH. A negative regulatory loop between microRNA and Hox gene controls posterior identities in Caenorhabditis elegans. PLOS Genetics. 2010; 6(9).
- Zhao, Z., Flibotte, S., Murray, J.I., Blick, D., Boyle, T.J., Gupta, B.P., Moerman, D.G., Waterston, R.H. New Tools for Investigating the Comparative Biology of Caenorhabditis briggsae and Caenorhabditis elegans. Genetics. 2010.
- Aydin, Z., Murray, J.I., Waterston, R.H., Noble, W.S. Using machine learning to speed up manual image annotation: application to a 3D imaging protocol for measuring single cell gene expression in the developing C. elegans embryo. BMC Bioinformatics. 2010.
- Liu, X., Long, F., Peng, H., Aerni, S.J., Jiang, M., Sanchez-Blanco, A., Murray, J.I., Waterston, R.H., Batzoglou, S., Myers, E.W., Kim, S.K. Analysis of gene regulation and cell fate from single-cell gene expression profiles in C. elegans. Cell. 2009.
- Murray, J.I., Bao, Z., Boyle, T.J., Boeck, M.E., Mericle, B., Nicholas, T.J., Zhao, Z., Sandel, M.J., Waterston, R.H. Automated analysis of embryonic gene expression with cellular resolution in C. elegans. Nature Methods. 2008.
- Yoshimura, S., Murray, J.I., Lu, Y., Waterston, R.H., Shaham, S. mls-2 and vab-3 control glia development, hlh-17/Olig expression and glia-dependent neurite extension in C. elegans. Development. 2008.
- Zhao, Z., Boyle, T.J., Bao, Z., Murray, J.I., Mericle, B., Waterston, R.H. Comparative analysis of embryonic cell lineage between Caenorhabditis briggsae and Caenorhabditis elegans. Developmental Biology. 2008.
- Bao, Z., Zhao, Z., Boyle, T.J., Murray, J.I., Waterston, R.H. Control of cell cycle timing during C. elegans embryogenesis. Developmental Biology. 2008.
- Jacobs, S.B., Basak, S., Murray, J.I., Pathak, M., Attardi, L.D. Siva is an apoptosis-selective p53 target gene important for neuronal cell death. Cell Death and Differentiation. 2007.
- Boyle, T.J., Bao, Z., Murray, J.I., Araya, C.L., Waterston, R.H. AceTree: a tool for visual analysis of Caenorhabditis elegans development. BMC Bioinformatics. 2006.
- Murray, J.I., Bao, Z., Boyle, T.J., Waterston, R.H. The lineageing of fluorescently-labeled Caenorhabditis elegans embryos with StarryNite and AceTree. Nature Protocols. 2006.
- Bao, Z., Murray, J.I., Boyle, T., Ooi, S.L., Sandel, M.J., Waterston, R.H. Automated cell lineage tracing in Caenorhabditis elegans. PNAS. 2006.
- Hillier, L.W., Coulson, A., Murray, J.I., Bao, Z., Sulston, J.E., Waterston, R.H. Genomics in C. elegans: so many genes, such a little worm. Genome Research. 2005.
- Murray, J.I., Whitfield, M.L.,Trinklein, N.D., Myers, R.M., Brown, P.O. and Botstein, D. Diverse gene expression responses to stress in human cell lines. Molecular Biology of the Cell. 2004.
- Trinklein, N.D., Murray, J.I., Hartman, S., Botstein, D. and Myers, R.M. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Molecular Biology of the Cell. 2004.
- Whitfield,M.L., Finlay,D., Murray,J.I., A.Troyanskaya,O., Chi,J.A., Brown,P.O., Botstein,D. and Connolly,M.K. Systemic and cell-type specific gene expression patterns in scleroderma skin. PNAS. 2003.
- Whitfield,M.L., Sherlock, G., Saldanha, A.,Murray, J.I., Ball, C.A., Alexander, K.E., Matese, J.C., Perou, C.M., Hurt, M.M., Brown, P.O., Botstein, D. Identification of Genes Periodically Expressed in the Human Cell Cycle and Their Expression in Tumors Molecular Biology of the Cell. 2002.