Murray Lab

Ensuring robust and precise development
The goal of the Murray laboratory is to understand how genomes control animal development. Animals, including humans, consist of many highly specialized cell types which must be generated in the correct number and at the right time and place to allow the organism to function correctly. Most of our understanding of this process comes from studies that focus on individual cell types or tissues, limiting our understanding of how regulators and mechanisms apply across all cells in an organism. The Murray laboratory develops and uses whole-organism live-cell imaging, and genomics methods, combined with classical genetics, to study gene regulation across the entire embryo. We use the nematode worm Caenorhabditis elegans as a model for many of our studies because it shares most of its cell types and regulatory mechanisms with humans, but is much easier to study.
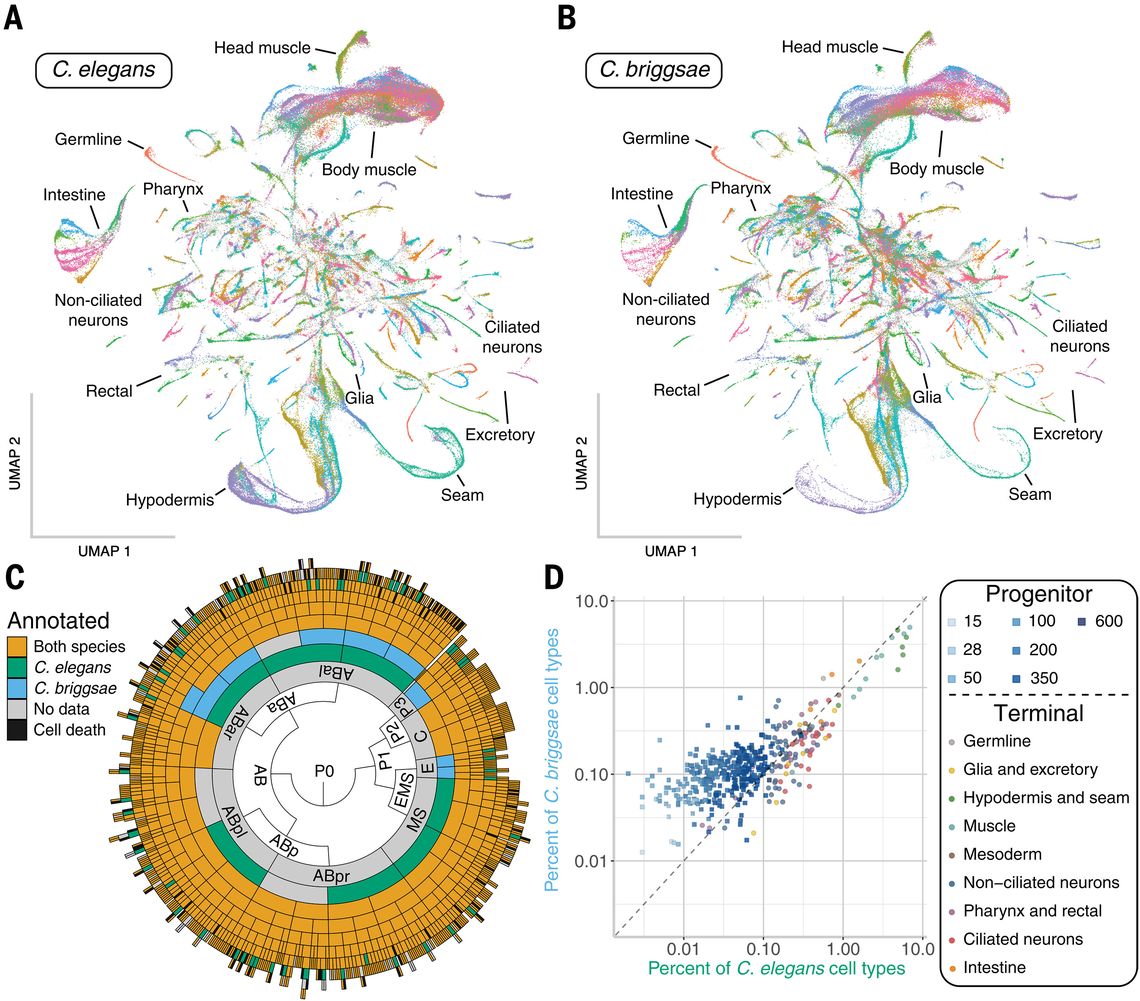
"Lineage-resolved analysis of embryonic gene expression evolution in C. elegans and C. briggsae"
Science 2025
Congratulations to Dr Felicia Peng for her paper and completing her PhD in the Murray Lab!!

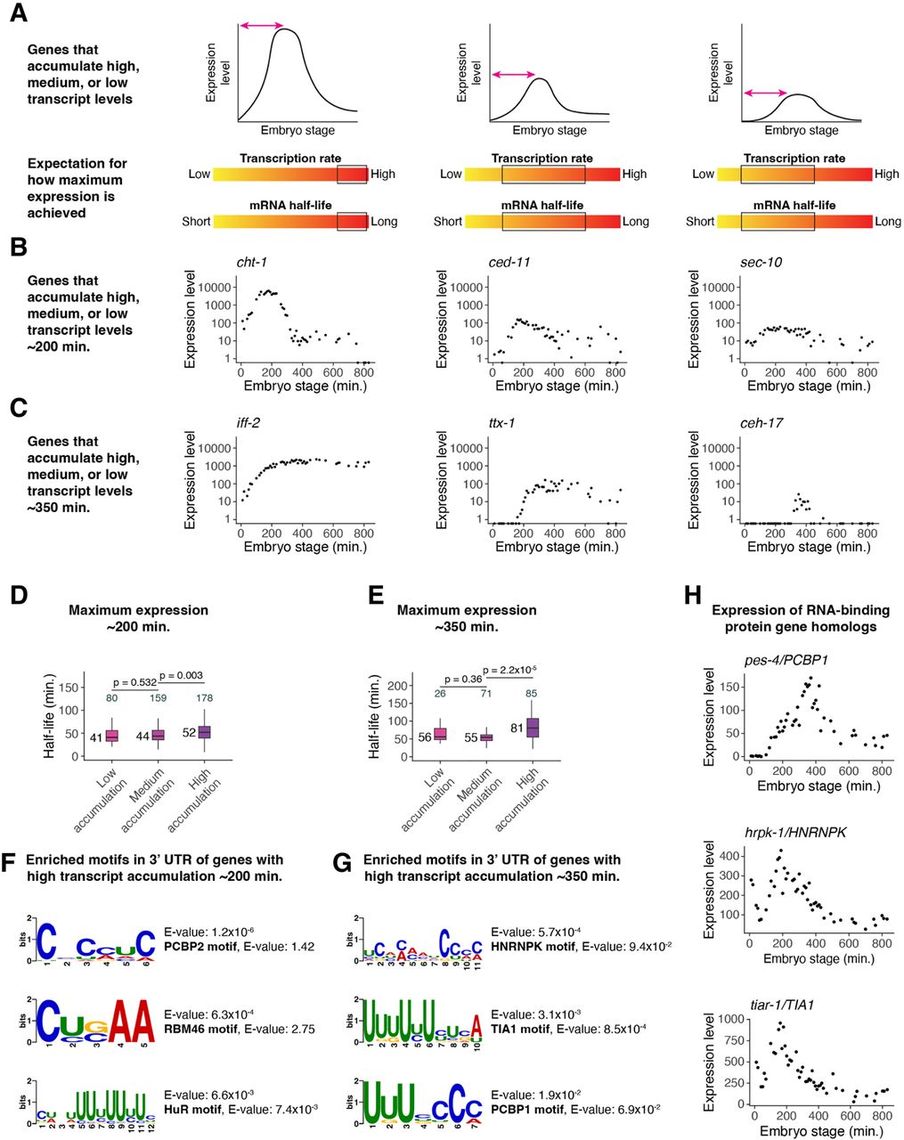
"A spatiotemporally resolved atlas of mRNA decay in the C. elegans embryo reveals differential regulation of mRNA stability across stages and cell types"
Genome Research 2024
Congratulations to Undergraduate graduates Jack Marchese & Erin Marble and to Masters graduate Yuntian Gan!



Congratulations on our post-doc alumnus Priya Sivaramakrishnan opening her own lab!
Dr Sivaramakrishnan's lab website
"Mechanisms of lineage specification in Caenorhabditis elegans."
Genetics 2023
Read this paper
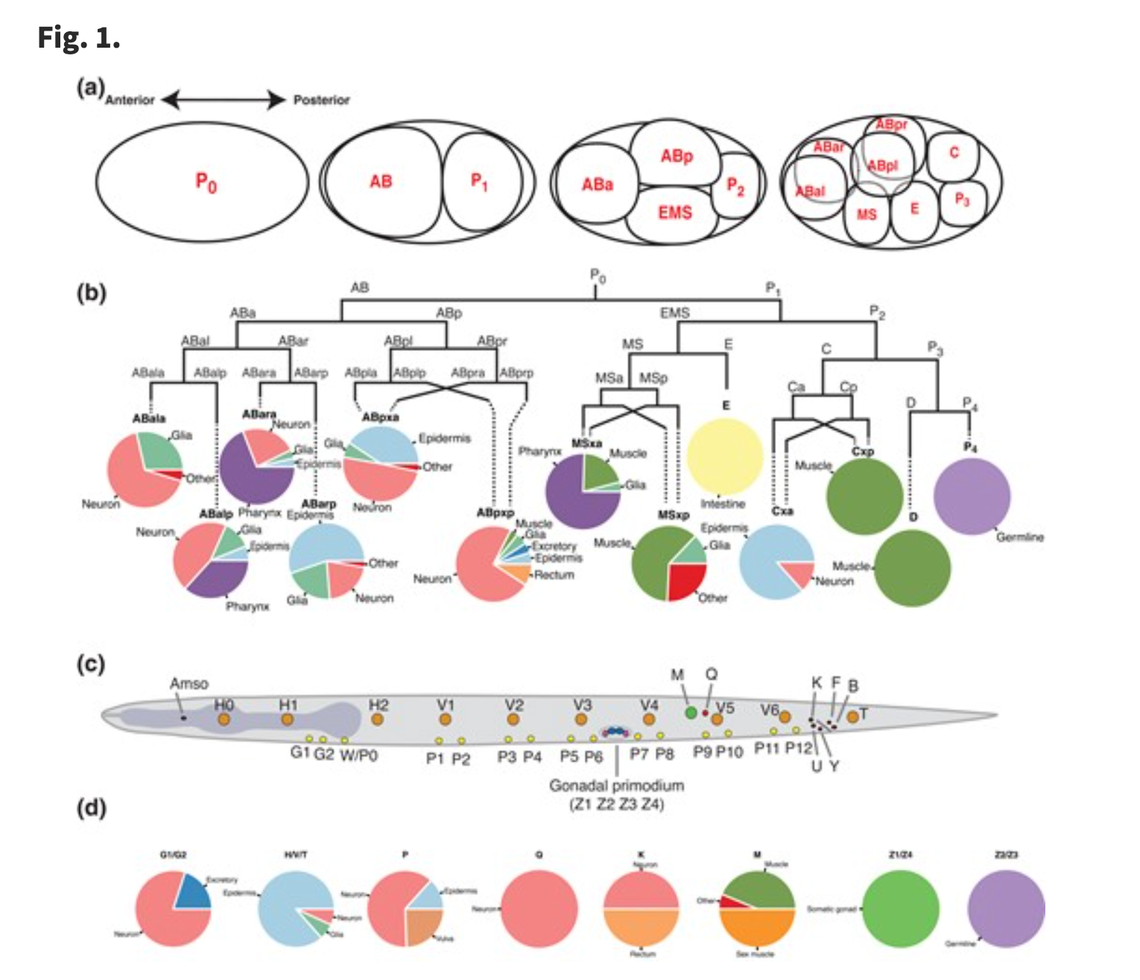
"A lineage-resolved molecular atlas of C. elegans embryogenesis at single-cell resolution."
Science 2019
Read this paper
Read an article about this paper
|
|
|
3D projection of single cell RNA expression data from C. elegans embryos |

RESULTS
We profiled the transcriptomes of 86,024 single cells from C. elegans embryos at developmental stages ranging from gastrulation to terminal cell differentiation. Using computational methods, gene expression patterns from the literature, and gene expression data obtained from three-dimensional (3D) movies of fluorescent reporter lines, we mapped each single-cell transcriptome to its corresponding position in the known C. elegans cell lineage tree. In total, we identified 502 distinct terminal and preterminal cell types, which correspond to 1068 individual branches of the lineage tree. We computed a transcriptional profile for each detected cell type and determined the gene expression differences between mother and daughter cells, and between sister cells, for >200 cell division events in the lineage.
Congratulations on our post-doc alumnus Amanda Zacharias opening her own lab!
Dr Zacharias's lab website
We value positivity and diversity and are a great lab for proactive and curious scientists.
Want to join us?
Contact John to apply for a postdoctoral position. We are affiliated with several graduate programs at Penn including:
- Biomedical Graduate Studies
- Cell and Molecular Biology/Genetics and Epigenetics
- Genomics and Computational Biology
- Biochemistry and Molecular Biophysics
- Bioengineering