Research Program
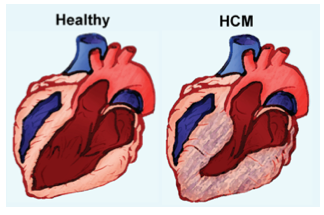
The Day lab is broadly interested in genetic heart muscle diseases. Our research program integrates translational and clinical science, with a primary focus on hypertrophic cardiomyopathy. We want to understand the full spectrum of disease evolution and progression, from the primary effects of sarcomere gene mutations in cardiac muscle cells to the phenotypic expression and predictors of adverse outcomes in patients. We have several avenues we are pursuing in laboratory-based research projects and also projects that analyze data collected from patient registries and data repositories.
Current Projects
Genetic variants in myosin binding protein C (MYBPC3) account for half of all cases of familial hypertrophic cardiomyopathy (HCM). MyBP-C (the protein encoded by MYBPC3) binds to myosin in the thick filament and actin in the thin filament to regulate contraction of the sarcomere. Most of the variants that cause HCM result in a premature termination codon and commonly referred to as “truncating” variants. A smaller but important subset of variants are non-truncating.
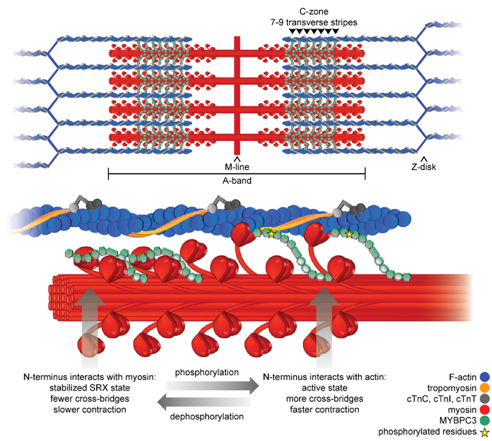
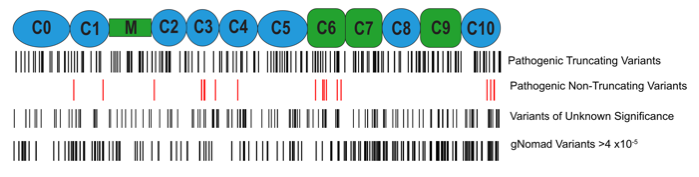
The truncating variants cause HCM because they result in haploinsufficiency, or a decreased in the amount of MyBP-C in the sarcomere. Some of the non-truncating variants also appear to act by this mechanism, while others presumably alter the way MyBP-C functions and interacts with other proteins. Our lab continues to be interested in how precisely these different variants cause HCM. A major current direction of our lab is exploring way in which haploinsufficiency could be corrected and the amount of MyBP-C normalized, in the presence of a truncating variant on one of the alleles. We have identified the molecular chaperone, HSP70, as a critical regulator of MyBP-C protein turnover. We have shown that HSP70 facilitates degradation of MyBP-C in cardiac myocytes. This chaperone exists in complex with >50 other co-chaperones, some of which confer specificity for specific protein substrates. We are interested in identifying which of the constituents of this complex are involved in degradation of MyBP-C, with the idea that we could develop pharmacologic therapies to interfere with this interaction and stabilize wild-type MyBP-C produced from the normal allele.
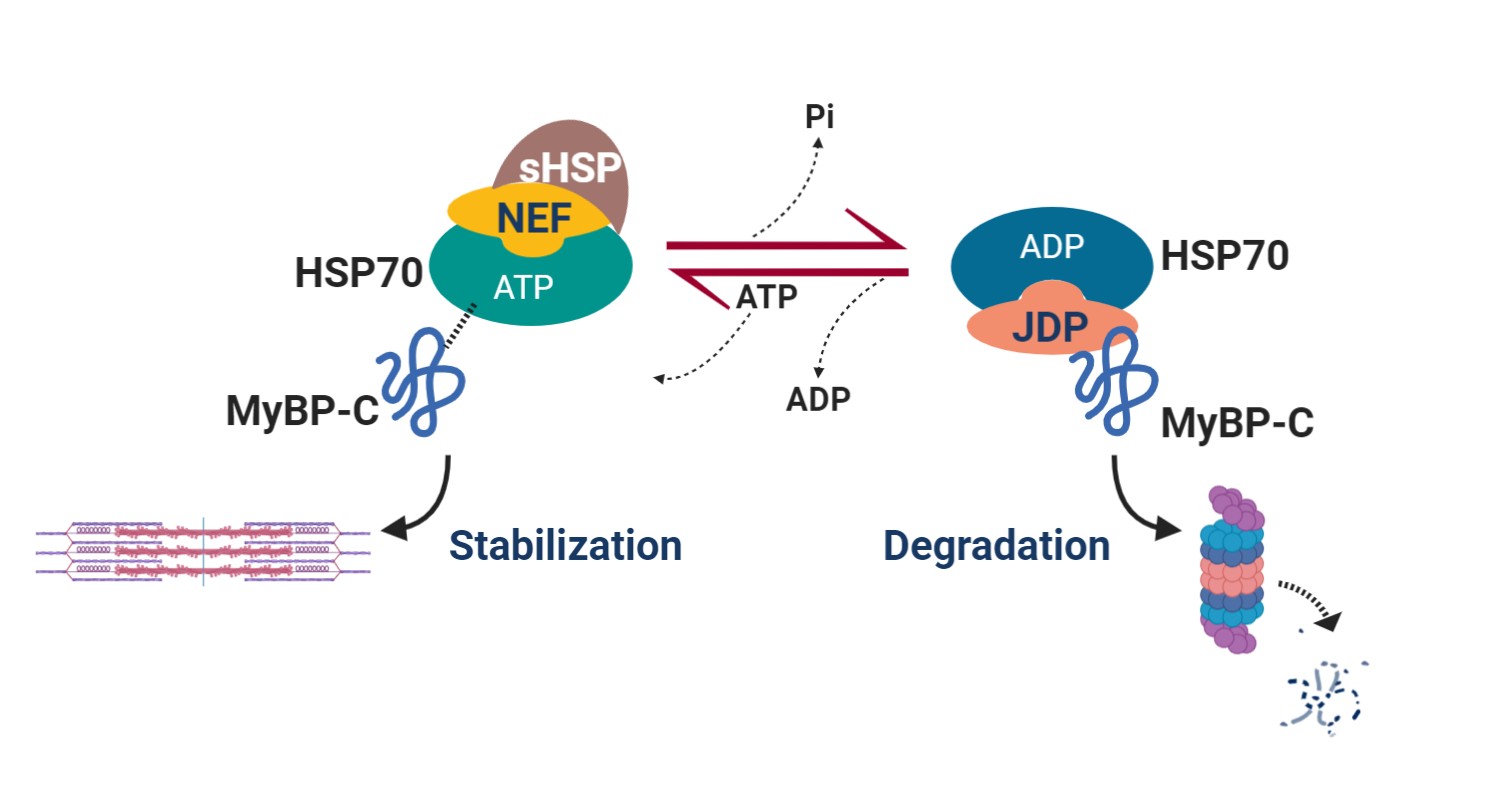
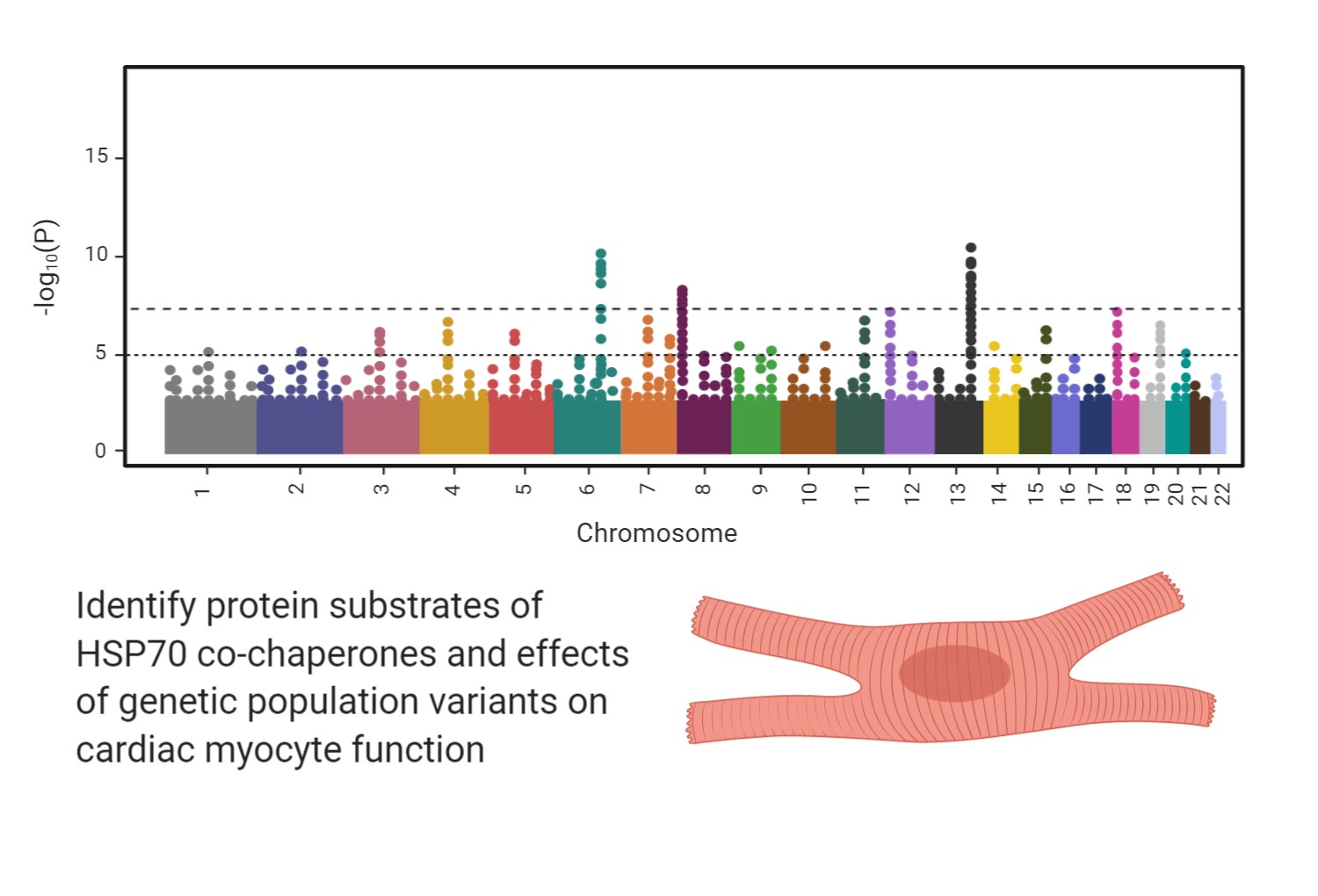
Common variants in several molecular chaperone genes have been shown to be associated with heart failure in large genome-wide association studies. We are taking a genome-first approach to identify additional chaperones and co-chaperones in the HSP70 complex that may be implicated in cardiomyopathy, heart failure, and cardiac arrhythmic phenotypes. We are also performing functional studies in the laboratory to define the underlying mechanisms behind these strong associations between chaperone genetic variation and heart failure.
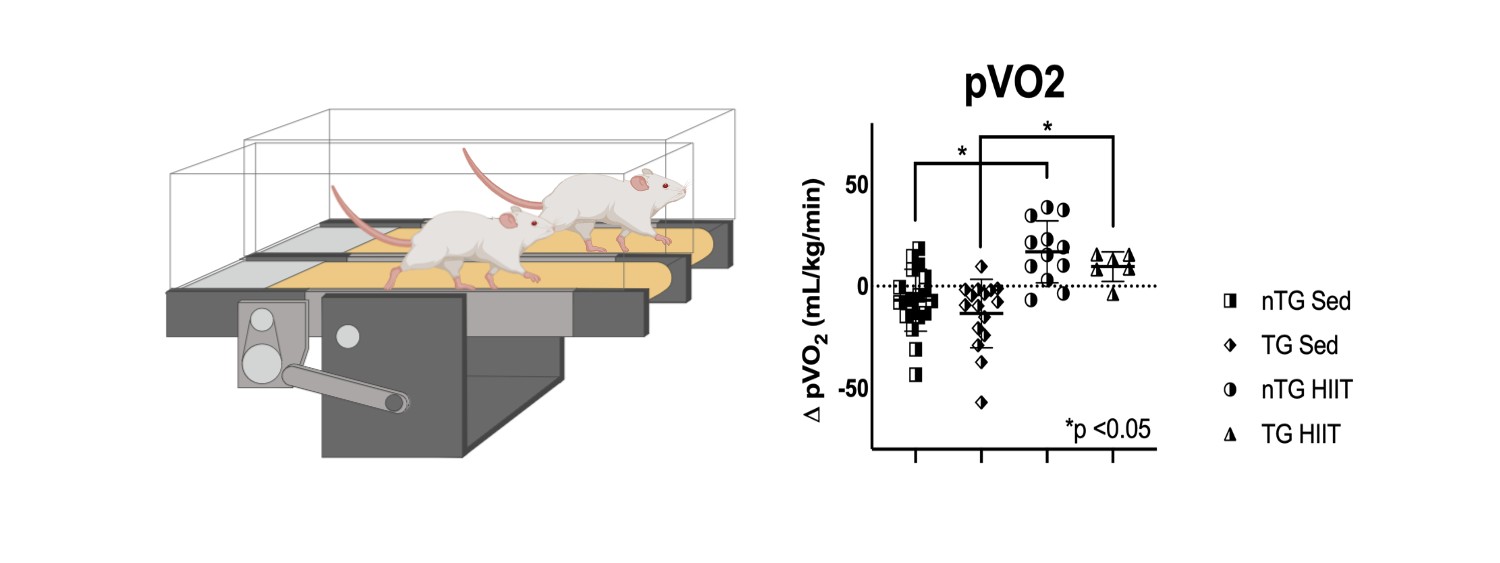
Exercise in HCM has been a major focus of both clinical and translational research for our group. In a previous randomized clinical trial in patients (RESET-HCM) we demonstrated that regular exercise improves cardiorespiratory fitness without any negative consequences. We are now focusing on maximizing that improvement through more intense exercise protocols in mice. These studies will also allow us to understand if and how exercise improves the adverse remodeling in HCM.
This is a new area of interest for our lab and one that benefits greatly from collaborations among other CVI investigators. Metabolic defects, including impaired fatty acid oxidation, are prevalent in hypertrophic cardiomyopathy and likely contribute to disease pathogenesis. Like other forms of cardiomyopathy that cause heart failure, metabolic modulation to improve substrate utilization and ATP production is a promising opportunity for therapeutic intervention.
Clinical Research
Dr. Day is a founding investigator of the Sarcomeric Human Cardiomyopathy Registry (SHaRe) which was formed in 2015. This is a multicenter, international registry of patients with hypertrophic cardiomyopathy, now totaling almost 10,000 patients. We continue to learn a lot about the natural history of HCM and have been able to identify genetic and other factors that predict how the disease course will progress.