Traumatic Brain Injury
Neurodegenerative Changes Following Traumatic Brain Injury
Traumatic brain injury (TBI) is one of the most devastating diseases due to its high percentage of mortality and disability and its claim of over 2 million victims each year in the United States alone. Recent studies have addressed that brain trauma leads to an increased risk of developing Alzheimer's disease (AD) and induces the acute formation of AD-like plaques containing amyloid-β (Aβ). However, the novel mechanisms of neurodegenerative processes following brain trauma have yet to be identified.
To further explore the potential link between brain trauma and neurodegeneration, researchers used experimental brain injury models in both the pig (by inducing coronal plane rotational acceleration 110°) and in the rat ( lateral fluid percussion injury) to identify acute and prolong neurodegenerative changes after injury. Both animal models can produce clinically relevant neuropathological sequelae in the brain. They have also used human brain material from autopsies to study the connection between traumatic brain injury and AD, and to confirm similar findings gained from animal studies.
Progressive axonal pathology after TBI Progressive axonal pathology after TBI
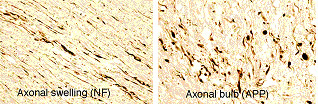
Traumatic axonal injury (TAI) is an important pathological finding. After rapid mechanical deformation of the brain during trauma, axonal cytoskeleton was damaged and axoplasmic transport was impaired. These processes cause the accumulation of transported proteins, including toxic proteins and peptides that can lead to secondary disconnection. Immunohistochemistry demonstrated high density of axonal swellings and axonal bulbs labeled by NF and APP antibodies persisted up to many years post-injury.
Axonal Aβ accumulation after TBI Axonal AΒ accumulation after TBI
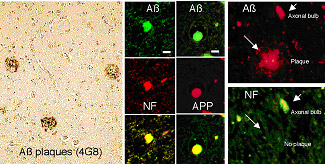
Following brain trauma, a marked accumulation of APP has been found in damaged axons. Furthermore, conversion of axonal APP to Aβ has been observed both clinically and in experimental models. Widespread axonal Aβ in the white matter and examples of axonal Aβ associated with Aβ plaques were confirmed by double immunostaining in brain-injured patients, the rat TBI model, and the swine model of DAI.
Aβ production mediated by β-secretase and presenilin after TBI Aβ production mediated by β-secretase and presenilin after TBI
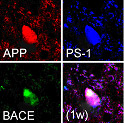
The primary mechanism for production of Aβ peptides is thought to be via transmembrane cleavage of APP by β-and g-secretases in AD. However, for brain trauma, the rapid production and aggregation of Aβ may result from unique mechanisms related to specific post-injury processes. Emerging data suggests that intra-axonal proteolysis of APP to Aβ is a primary process proposed for Aβ formation in traumatic brain injury. A recent study has found that Aβ production was mediated by beta-site APP cleaving enzyme (BACE) and the catalytic component of g -secretase, presenilin-1 (PS-1), within the axon membrane compartment of peripheral nerves. Further, it was demonstrated that APP functions as a kinesin-1 membrane receptor, mediating fast axonal transport of BACE and PS-1. Researchers found that the co-accumulation of Aβ with APP, BACE or PS-1in damaged axons in both acute and long-term survival human and animal models after brain trauma. These data support the hypothesis that trauma impairs axonal transport and leads to massive accumulation of several key axonal proteins that normally do not encounter each other in such high concentrations. The metabolic mechanism of Aβ formation in TBI shares similarities with Aβ formation in AD.