Essential Documents
Essential documents which include regulatory binders (also known as Study Admin File, Investigator Binder, Investigational Site File (ISF), or Study Binder) contain the study specific information and regulatory documentation in a centralized location. It organizes essential study documents and should be easy to access and auditable.
Site Regulatory Binder Essentials
-
Correspondence
- IRB
- Penn Reviewing Entities
- Sponsor/CRO
- General
- Study Documents
- Protocol/Protocol Amendments and supporting documents
- Informed Consent & HIPAA Authorization
- Case Report Form Sample
- Investigator’s Brochure /Device Manual
- FDA Forms as applicable – 1572, sample of labels, FDA correspondence, financial disclosures
- Lab Documents (CLIA/CAP)
- Study Tracking Logs
- Subject Logs (Screening/Enrollment)
- Adverse Event Reports
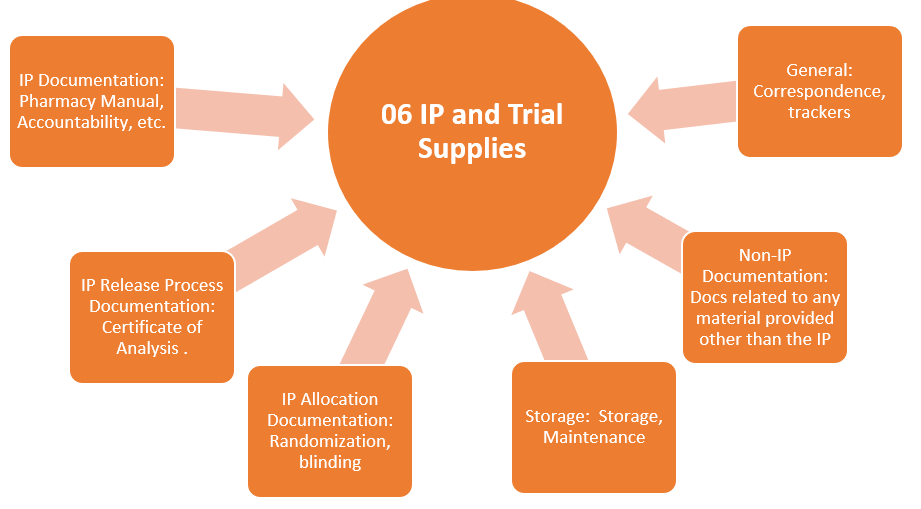
- Investigational Product
- Monitoring Visits and Logs
- Research Personnel
- Study personnel (CV, licenses, training, etc.)
- Delegation of Authority Log
- Other
- Unmasking Procedures for Blinded Trials
- Certificates of Confidentiality
- Other study documents as needed
It is preferable that the essential documents are maintained electronically, either through Penn Box or Veeva Vault, which both serve as options for electronic Regulatory Binders. Contact OCR for an eTemplate for maintaining the Regulatory Binder in PennBox.
OCR offers downloadable templates with instructions for maintaining regulatory binders on paper.
There are three different templates available within our forms, tools and templates library depending on the study type:
- Study Admin File_Device Template for developing a study administration file or regulatory binder for a device trial.
- Study Admin File_Social Behavioral Template for developing a study administration file or regulatory binder for a social behavioral study.
- Study Admin File_Drug Template for developing a study administration file or regulatory binder for a drug trial.
Sponsor Trial Master File (TMF)
The sponsor essential documents are filed in what is known as the Trial Master File (TMF). It should begin at first inception of a trial and should be maintained current and complete during the life cycle of the investigation until its completion.
For some studies, an individual might be both the PI and the (Regulatory) Sponsor. In this case, both sets of responsibilities mentioned apply.
The table below lists examples of essential documents maintained exclusively by the Sponsor in the TMF, maintained exclusively by the PI in the Reg. Binder and maintained by both.
| Examples of Sponsor Docs for Sponsor TMF only | Examples of Docs Shared by Both | Examples of PI Docs for Reg Binder only |
|---|---|---|
| Sample of Label(s) | Investigator's Brochure and updates | Ad(s) for Subject Recruitment |
| Certificate(s) of Analysis of Investigational Products(s) Shipped | Signed Protocol and Amendments and Sample Case Report Form (CRF) | Signed Informed Consent Forms |
| Master Randomization List |
Original document and Any Revision To:
|
Source Documents |
|
Pre-Trial Monitoring Report |
Insurance Statement (Where Required) |
Subject Identification Code List |
|
Certificate(s) of Analysis |
Signed Agreement Between Involved Parties |
Subject Enrolment Log |
|
Monitoring Visit Reports |
IRB approvals/acknowledgements |
Completed Subject Identification Code List |
|
Audit Certificate (If Available) |
Institutional Review Board/Independent Ethics Committee Composition |
Final Report By Investigator To IRB/IEC Where Required, and Where Applicable, To The Regulatory Authority(ies) |
|
Final Trial Close-Out Monitoring Report |
Regulatory Authority(ies) Authorization/Approval/Notification of Protocol (Where Required), Protocol Amendment(s) and Other Documents |
|
|
Treatment Allocation and Decoding Documentation |
CV and/or Other Relevant Documents Evidencing Qualifications of Investigator(s) and Subinvestigator(s) |
|
| Normal Value(s)/Range(s) and Updates For Medical/Laboratory/Technical Procedures(s) and/or Test(s) Included In The Protocol | ||
|
Medical/Laboratory/Technical Procedures/Tests and Updates
|
||
|
Instructions For Handling of Investigational Product(s) and Trial-Related Materials |
||
|
Shipping Records For Investigational Product(s) and Trial-Related Materials |
||
|
Decoding Procedures For Blinded Trials |
||
|
Instructions For Handling of Investigational Product(s) and Trial-Related Materials |
||
|
Shipping Records For Investigational Product(s) and Trial-Related Materials |
||
|
Decoding Procedures For Blinded Trials |
||
|
Trial Initiation Monitoring Report |
||
|
Documentation of Investigational Product(s) and Trial-Related Materials Shipment |
||
|
Relevant Communications Other Than Site Visits
|
||
|
Signed, Dated, and Completed Case Report Forms (CRF) |
||
|
Documentation of CRF Corrections |
||
|
Notification By Originating Investigator To Sponsor of Serious Adverse Events and Related Reports |
||
|
Notification By Sponsor and/or Investigator To Regulatory Authority(ies) and IRB(s)/IEC(s) of Unexpected Serious Adverse Drug Reactions and of Other Safety Information |
||
|
Notification By Sponsor To Investigators of Safety Information |
||
|
Interim Or Annual Reports To IRB/IEC and Authority(ies) |
||
|
Subject Screening Log |
||
|
Signature Sheet |
||
|
Record of Retained Body Fluids/Tissue Samples (If Any) |
||
|
Investigational Product(s) Accountability At Site |
||
|
Documentation of Investigational Product Destruction |
||
|
Clinical Study Report |
Additionally,
- If the study data supports a marketing application, the records must be maintained for 2 years after a marketing application is approved by the FDA.
- If the study data does not support a marketing application, the records must be maintained for 2 years after shipment and delivery of the drug for the investigational use is discontinued and the FDA has been so notified.
- Certain documents might need to be retained for a longer period of time based on the Penn’s Research Administration Records Policy.
Access to the TMF must be controlled and limited to only to the study sponsor and sponsor-authorized individuals.
Standard procedures and naming conventions should be developed to ensure an easy location of the records.
OCR has adopted the CDISC TMF Reference Model, endorsed by the Drug Information Association (DIA), to organize the TMF documents and recommends Penn Box or Veeva eTMF as the platforms for maintaining your TMF. Additional information about using the Veeva eTMF (including access and training) may be found here.
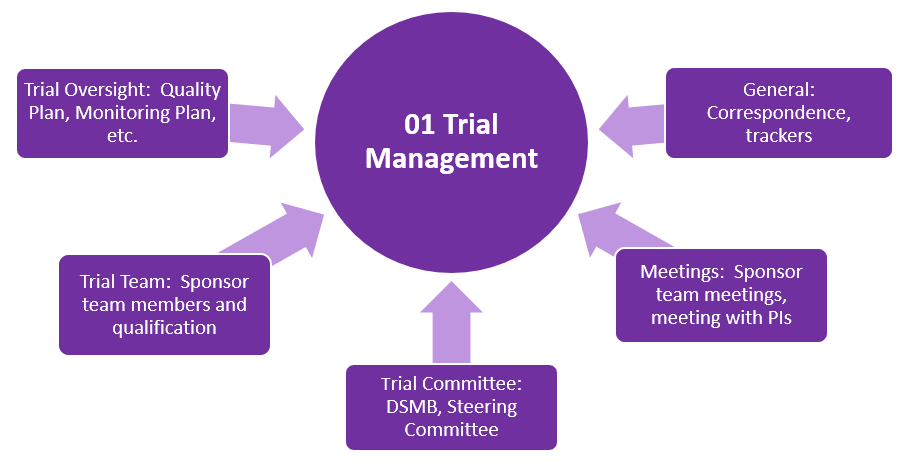
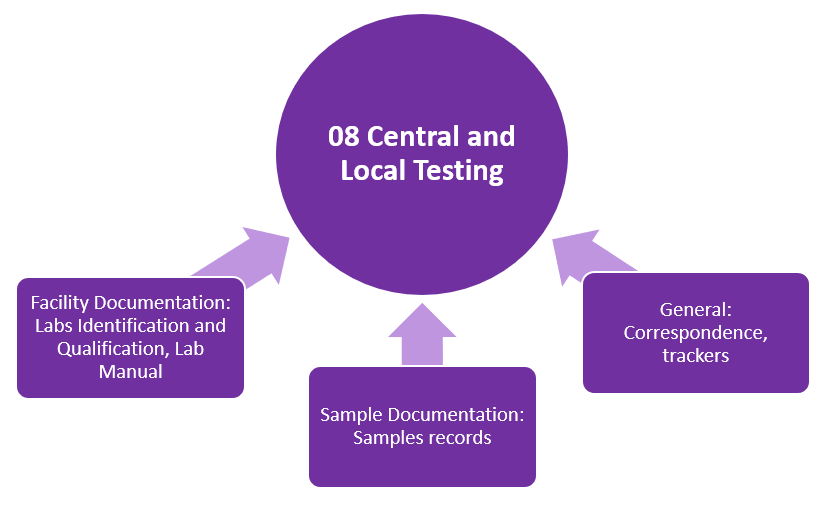
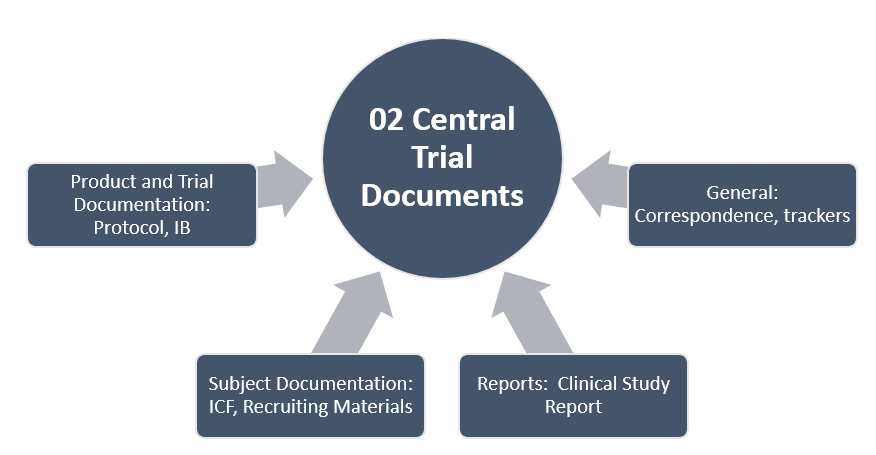
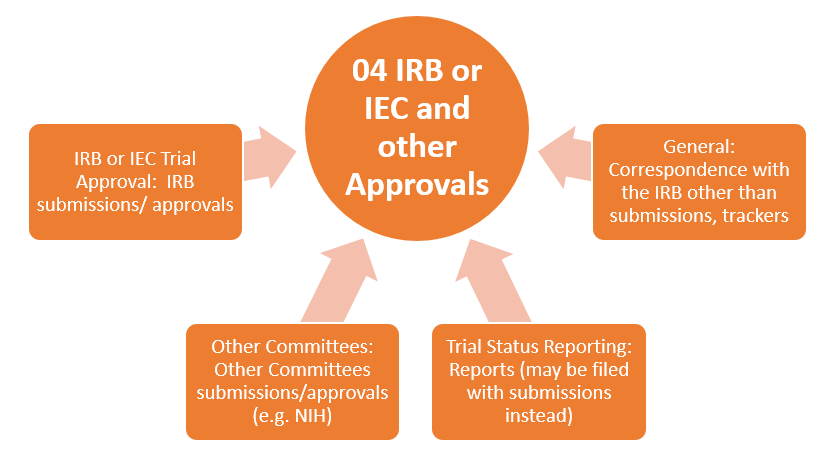
TMF Zones
This structure organizes the essential documents in eleven zones (categories). Each TMF zone is represented with the following graphics:
- The purple graphics represent zones containing documents generated by the Sponsor when setting up a trial.
- The gray graphics represent zones containing core documents necessary for the conduct of the study.
- The orange graphics represent zones containing documents generated by PI activities.
Contact OCR Regulatory for questions or assistance at PSOM-IND-IDE@pobox.upenn.edu