Research
We use novel animal (mouse) models, cultured cells, and human samples to tease out pathways and mediators involved in these processes. There are several interrelated projects currently focused on the following themes:
1. Novel Models and Mechanisms of Lung Fibrosis
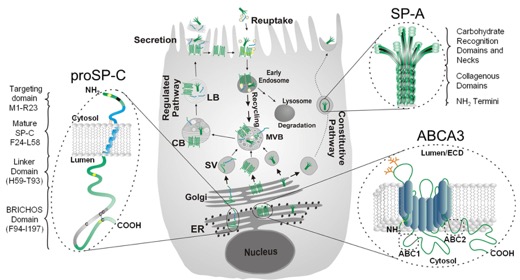
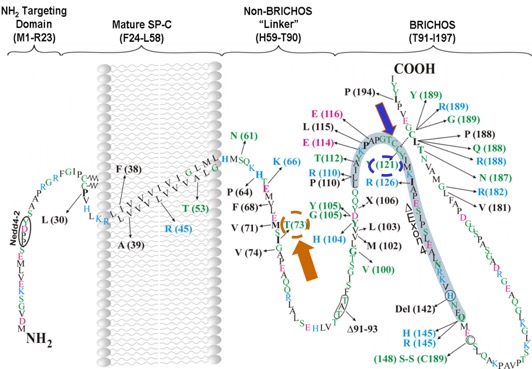
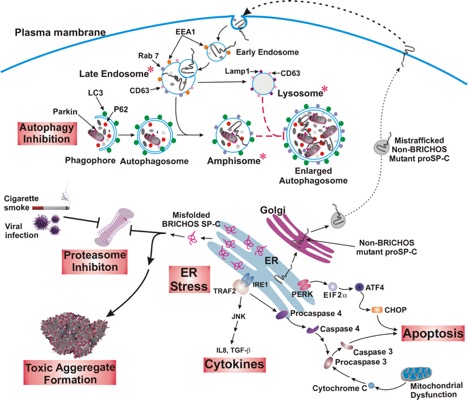
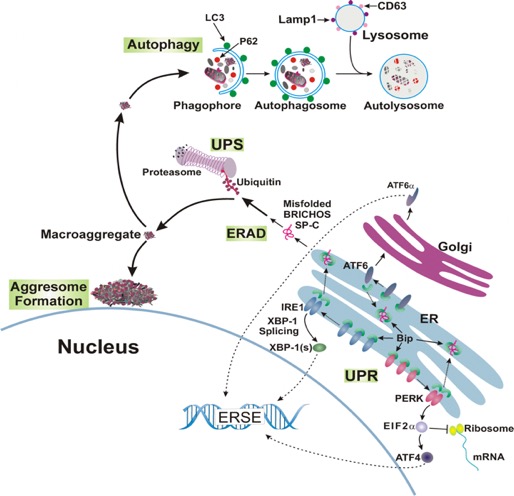
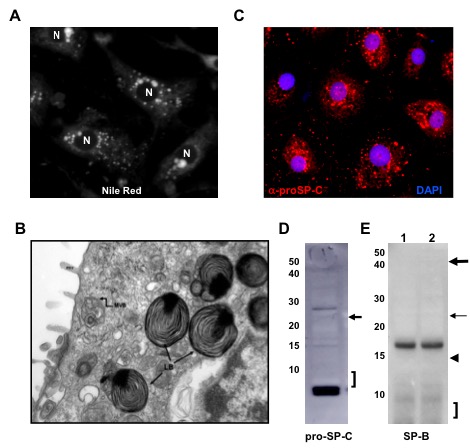
Mutations in some genes of the surfactant system have been associated with inherited forms of pulmonary fibrosis. We have shown that alterations in the proSP-C sequence that result in either misfolding or mistargeting of SP-C induce ER retention, formation of intracellular aggregates, or accumulation in plasma membrane and endosomes. Coincident with these observations, interstitial lung disease (ILD) in association with over 50 different heterozygous mutations in the SFTPC gene. Most mutations fall into 2 domains:
-
BRICHOS
- Aggregation prone mutants most commonly are found in a region of SFTPC with structural homology with a protein associated with familial Alzheimer-like dementia (termed BRI domain).
-
Non-BRICHOS
- Mutants which are diverted away normal anterograde trafficking to the lamellar body accumulate on the plasma membrane and in endosomal compartments.
We are using these "rare" genetic variants to understand the broader question of how lung fibrosis develops and progresses in sporadic (non-inherited) forms of the disease. Using Surfactant Protein C (SP-C) as a model substrate, we are characterizing the mechanisms underlying the development of epithelial cellular dysfunction and relating these back to the development of pulmonary fibrosis in vivo using primary human cells and mice expressing disease causing SP-C mutations. [See Figure 2, Figure 3, Figure 4, and Figure 5]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2. Epithelial Cell Dysfunction in the Pathogenesis of Pulmonary Fibrosis
In addition to disease causing variants in the SFTPC gene, we have explored the effects of mutations in other surfactant components (SP-A; ABCA3) as well as studied other monogenetic diseases that specifically affect lung epithelial cell function and have been associated with the development of pulmonary fibrosis. Current studies are directed at defining metabolic re-programming, alterations in macroautophagy, defective proteostasis and aging as drivers of dysfunctional AT2 phenotypes including aberrant basaloid cells.
3. Fibroblast Heterogeneity and Dynamics in the Pathogenesis of Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic lung disease characterized by injury to and/or dysfunction of the alveolar epithelium. Interactions between the alveolar epithelial type II cells (AT2) and the different fibroblast subpopulations require a tight regulation to maintain and regenerate the alveolar niche. Advances in single cell RNA-sequencing (scRNA-seq) have allowed the transcriptomic characterization of a unique Transitional/Inflammatory fibroblast population that emerge following injury. The role of this population and its interaction with AT2 to effect injury / repair of the alveolar niche has not been well described.
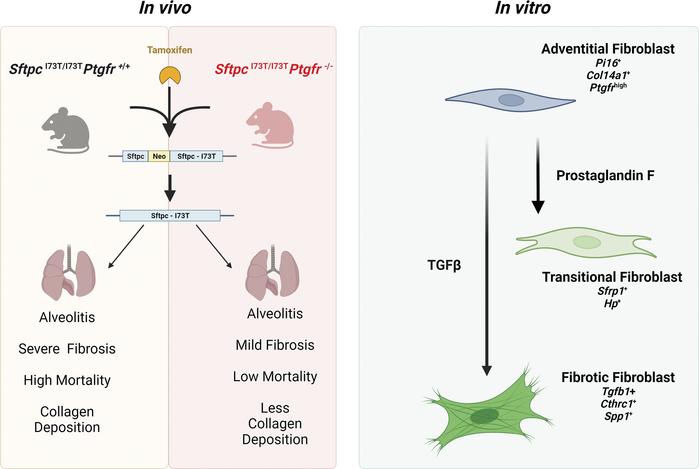
We use our novel SftpcI73T mice in which tamoxifen administration initiates an early multiphasic alveolitis through day 14 before transitioning to spontaneous fibrotic modeling by day 28. We have generated large scRNA-seq data sets collected from lungs at peak injury (day 14) and fibrotic remodeling (28) and after treatment with antifibrotic therapies. We define mesenchymal populations using marker genes from multiple published studies. Signaling crosstalk between different clusters and the epithelia is done using Cellchat and RNAscope to spatially localize these populations within the niche. We have in hand sorting strategies to isolate all major fibroblast populations (Alveolar, Adventitial and Transitional/Inflammatory, and CTHRC1(myo)fibroblasts) for use in organoid culture systems to characterize the epithelial-fibroblast crosstalk during the alveolar niche regeneration. This work has created a working model for fibroblast dynamics in lung injury/repair. [See Figure 8]
{kind=link}
4. The Role of Inflammatory Effector Cells and Tregs in Lung Injury / Repair
The phenotype of the mutant SFTPC mouse models includes an early inflammatory phase and a late fibrotic phase. Using high resolution techniques (FACS, RNA-seq, etc.) we are characterizing the role of monocyte-macrophage effector cells and more recently Foxp3-regulatory T cells, the signaling pathways that drive their recruitment, and their crosstalk with structural cells in the alveolar niche to better understand the pathogenesis of pulmonary fibrosis. [See Figure 6]
{kind=link}
5. AT2 Cell Biosynthetic and Quality Pathways in Health and Disease
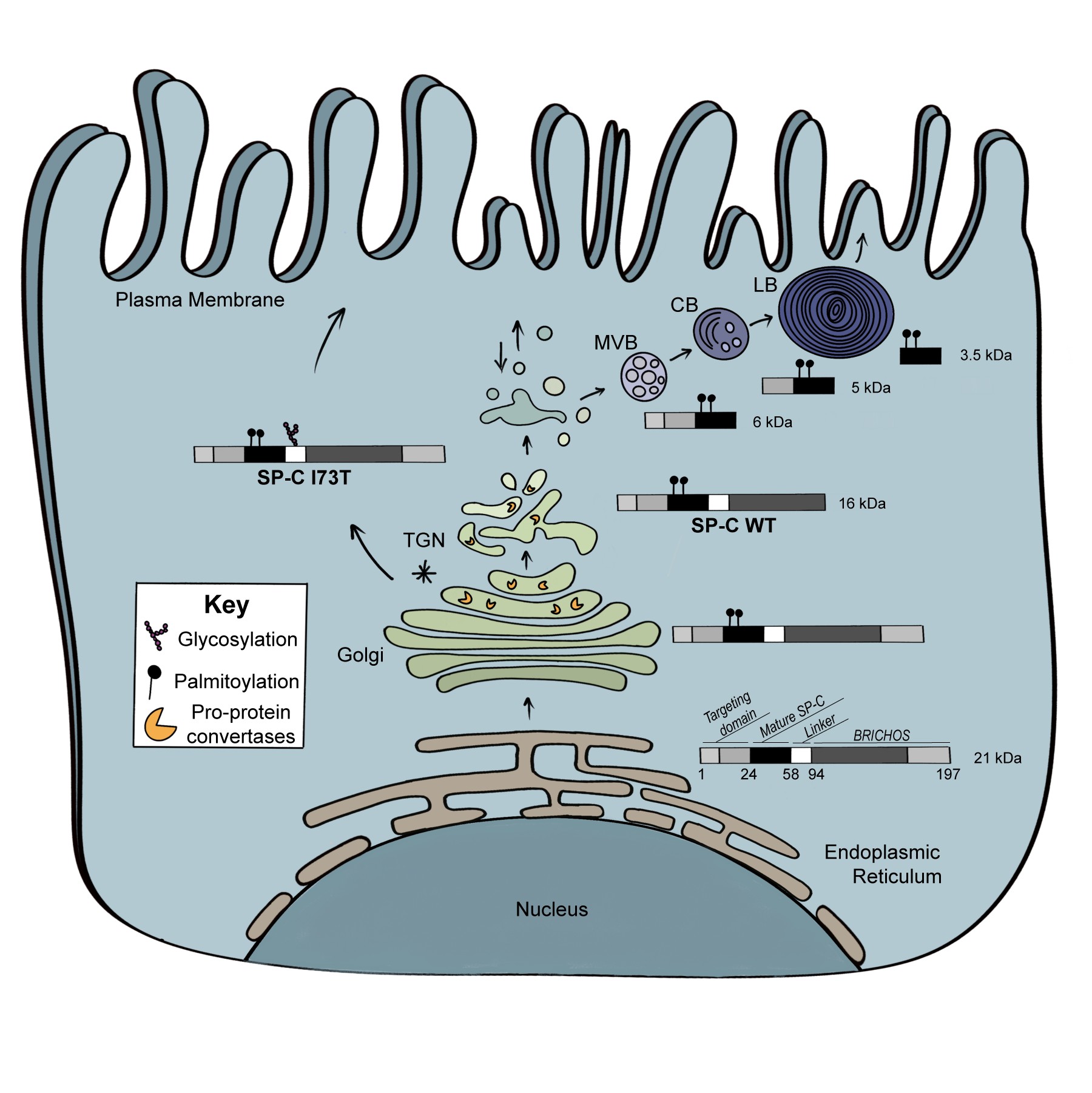
In a project spanning 25 years, we have been defining the cellular metabolism of pulmonary surfactant components including all 4 surfactant proteins with a current emphasis on Surfactant Protein C and the regulation of its intracellular trafficking including sorting in the Golgi and cleavage by Furin like proteases.
Cells utilize a variety of mechanisms and organelles to ensure proper synthesis of correctly folded proteins. We are characterizing these "quality control " mechanisms in alveolar type 2 cells using “SFTPC BRICHOS mutations” as substrates. These proteins can induce cell dysfunction (Figure 3) including formation of protein aggregates, generation of inflammation, induction of apoptosis, and changes to a profibrotic/senescent state. By continuing to leverage an extensive discovery science approach, we continue to identify new mechanisms of AT2 cell dysfunction in a variety of rare diseases including Hermansky-Pudlak Syndrome (HPS), Niemann-Pick disease, and COPA syndrome. [See Figure 1 and Figure 7]
{kind=link}
{kind=link}