Basic/Translational
Basic/Translational Researchers
- Caroline Bartman, PhD
- Gregory L. Beatty, MD, PhD
- Donita C. Brady, PhD
- Erica L. Carpenter, MBA, PhD
- Crystal S. Conn, PhD
- Constantinos Koumenis, PhD
- Ellen Puré, PhD
- Sydney M. Shaffer MD, PhD
- M. Celeste Simon, PhD
- Ben Z. Stanger, MD, PhD
- Ioannis Verginadis, MSc, PhD
- Robert H. Vonderheide, MD, DPhil
- Kathryn E. Wellen, PhD
- Kenneth S. Zaret, PhD
- Rong Zhou, PhD
Caroline Bartman, PhD
The Bartman laboratory aims to identify metabolic pathways altered in tumor cells, or in tumor-resident immune cells, and target those pathways to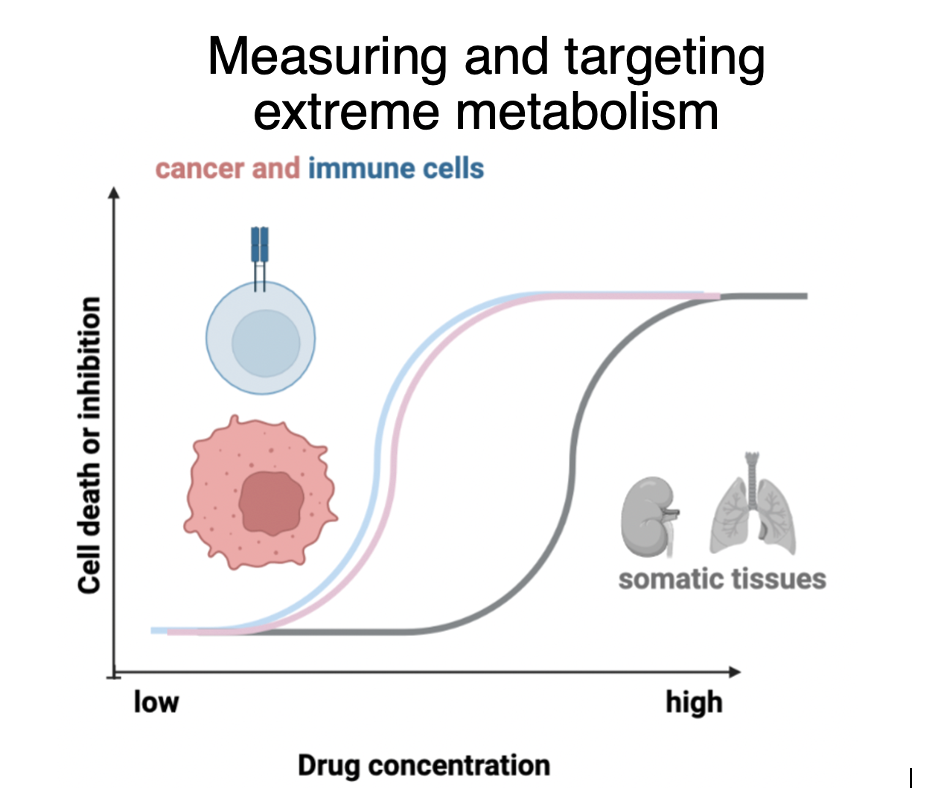 treat cancer. We measure tumor metabolic fluxes in vivo using carbon-13-labeled non-radioactive nutrient infusions, read out by liquid chromatography-mass spectrometry metabolomics. We developed techniques to measure glycolysis and the Krebs cycle in vivo. Using these approaches, we found that primary solid tumors make and use energy slower than most healthy tissues: tumors exhibit slow Krebs cycle flux and fail to upregulate glycolysis enough to compensate in terms of ATP production. In future, we aim to measure the metabolism of immune cells in tumors, especially myeloid cells in pancreatic cancer. We also will develop techniques to measure other key metabolic fluxes in tumors, such as glycan synthesis pathways. Finally, we will test strategies to manipulate immune cell metabolism to boost antitumor immunity, such as engineering T cells to store up glycogen ex vivo to enable their nutrient status, tumor entry and function.
treat cancer. We measure tumor metabolic fluxes in vivo using carbon-13-labeled non-radioactive nutrient infusions, read out by liquid chromatography-mass spectrometry metabolomics. We developed techniques to measure glycolysis and the Krebs cycle in vivo. Using these approaches, we found that primary solid tumors make and use energy slower than most healthy tissues: tumors exhibit slow Krebs cycle flux and fail to upregulate glycolysis enough to compensate in terms of ATP production. In future, we aim to measure the metabolism of immune cells in tumors, especially myeloid cells in pancreatic cancer. We also will develop techniques to measure other key metabolic fluxes in tumors, such as glycan synthesis pathways. Finally, we will test strategies to manipulate immune cell metabolism to boost antitumor immunity, such as engineering T cells to store up glycogen ex vivo to enable their nutrient status, tumor entry and function.
Gregory L. Beatty, MD, PhD
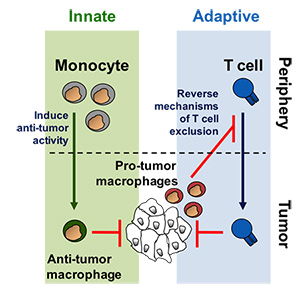
Dr. Beatty's laboratory incorporates both basic science research and clinical investigation to examine the role of innate immunity, in particular monocyte/macrophages, in regulating tumor biology and T cell immune privilege in pancreatic ductal adenocarcinoma (PDAC). Monocytes/macrophages are continually recruited to PDAC lesions and most often support tumor growth, invasion and metastasis, whereas effector T cells are rarely observed to infiltrate tumors. The exclusion of T cells from PDAC remains a critical barrier to the successful translation of novel immunotherapies for this disease. The Beatty laboratory is employing genetic mouse models of PDAC in combination with early phase clinical trials (including adoptive cellular therapy and novel immunomodulatory drugs) to understand mechanisms that 1) determine pro- versus anti-tumor activity of myeloid cells recruited to primary and metastatic lesions and 2) regulate T cell exclusion from tumor tissue. The overall goal is to develop novel immune-based strategies that harness the anti-tumor potential of both myeloid and T cells to treat patients with PDAC.
Donita C. Brady, Ph.D.
The research interests of the Brady laboratory lie at the intersection of cancer biology, signal transduction,and transition metal homeostasis. Our lab seeks to define kinase signal transduction pathways downstreamof oncogenes that are allosterically modulated by transitional metals. Findings from these studies have thepotential to be leveraged as a means to perturb metal availability to essential kinase signaling cascades intumors. Namely, despite diverse risk factors, comprehensive whole exome sequencing has revealed that~95% of PDAC patients can be genetically characterized by a signature genetic event in which the smallGTPase KRAS is mutated to remain in an active oncogenic state. However, no effective anti-RAS therapieshave transitioned into the clinic for PDAC despite the ubiquity of KRAS mutations. Thus, the identificationof novel molecularly targetable vulnerabilities driven by oncogenic KRAS that contribute to PDAC is criticalfor combatting this disease. Many groups have demonstrated the mechanisms through which oncogenicKRAS-mediated metabolic reprogramming sustains unrestricted tumor growth, survival, and therapeuticresistance. One potent mechanism engaged by oncogenic KRAS is the constitutive induction of autophagy, which salvages building blocks for macromolecular biosynthesis and bioenergetics that are scarce due to the nutrient- and oxygen-poor tumor microenvironment of PDAC.
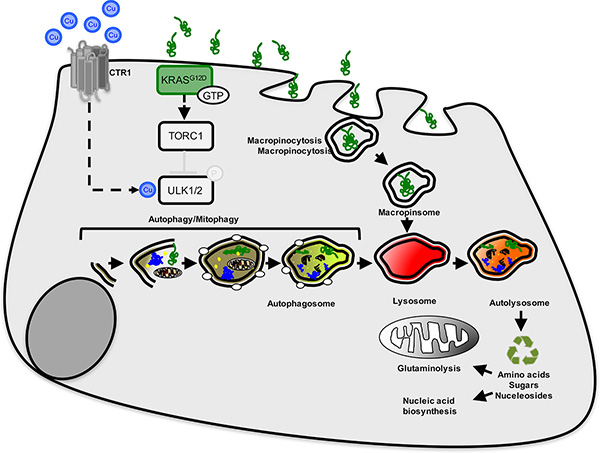
Molecularly the ability of cells to initiate autophagy is governed by kinase signaling networks that senseamino acid deprivation and low cellular ATP levels. Work by our group and others is beginning to uncovernovel functions for copper (Cu) as a non-structural intracellular mediator of signaling in the context of normalphysiology as well as the pathophysiology of diseases such as cancer. Preliminary studies from our labfound that the autophagy regulatory kinases, ULK1 and ULK2, bind to Cu and that Cu chelators can inhibitULK1 and ULK2 kinase activity. Moreover, decreasing Cu influx reduces basal and nutrient deprivedautophagy induction and decreases ULK1 and ULK2 activity. Thus, the goal of our research this is to buildon these promising preliminary data to: a) elucidate the contribution of Cu to autophagy induction throughdirect regulation of the ULK1 and ULK2 kinases and b) determine whether this unique signaling paradigmcan be exploited therapeutically via Cu reducing drugs to treat KRAS-driven PDAC.
Erica L. Carpenter, MBA, PhD
The Liquid Biopsy Laboratory, led by Director Dr. Erica Carpenter, focuses on the identification, capture, and analysis of Circulating Tumor Cells (CTCs), Disseminated Tumor Cells (DTCs), cell-free DNA (cfDNA), and exosomes from cancer patients, including those with pancreatic cancer. Blood, bone marrow, pleural effusions, and other non-invasively captured patient samples are used to detect biomarkers which allow: 1) early detection of disease as well as post-therapy monitoring of minimal residual disease, 2) an efficient means of determining clinical and biological response to therapy and, thus, clinical decision making, and, 3) cancer genetic phenotyping to drive personalized medicine that obviates the need for serial biopsies in a population of patients for which these procedures are difficult, risky, and insufficient.
The focus of the Liquid Biopsy Laboratory is driven by the needs of clinicians and translational investigators, and realized through collaborative work with investigators in the Penn School of Medicine, the Penn School of Engineering, and the Center for Personalized Diagnostics. Moreover, when it is determined that outsourcing of technology development is preferable, collaborative efforts with industry partners are actively sought, and these efforts have already been initiated in focused areas. In the case of pancreatic cancer, Dr. Carpenter’s lab focuses on studying early stages of human pancreatic cancer and cancer progression in collaboration with academic and industry partners.
Crystal S. Conn, PhD
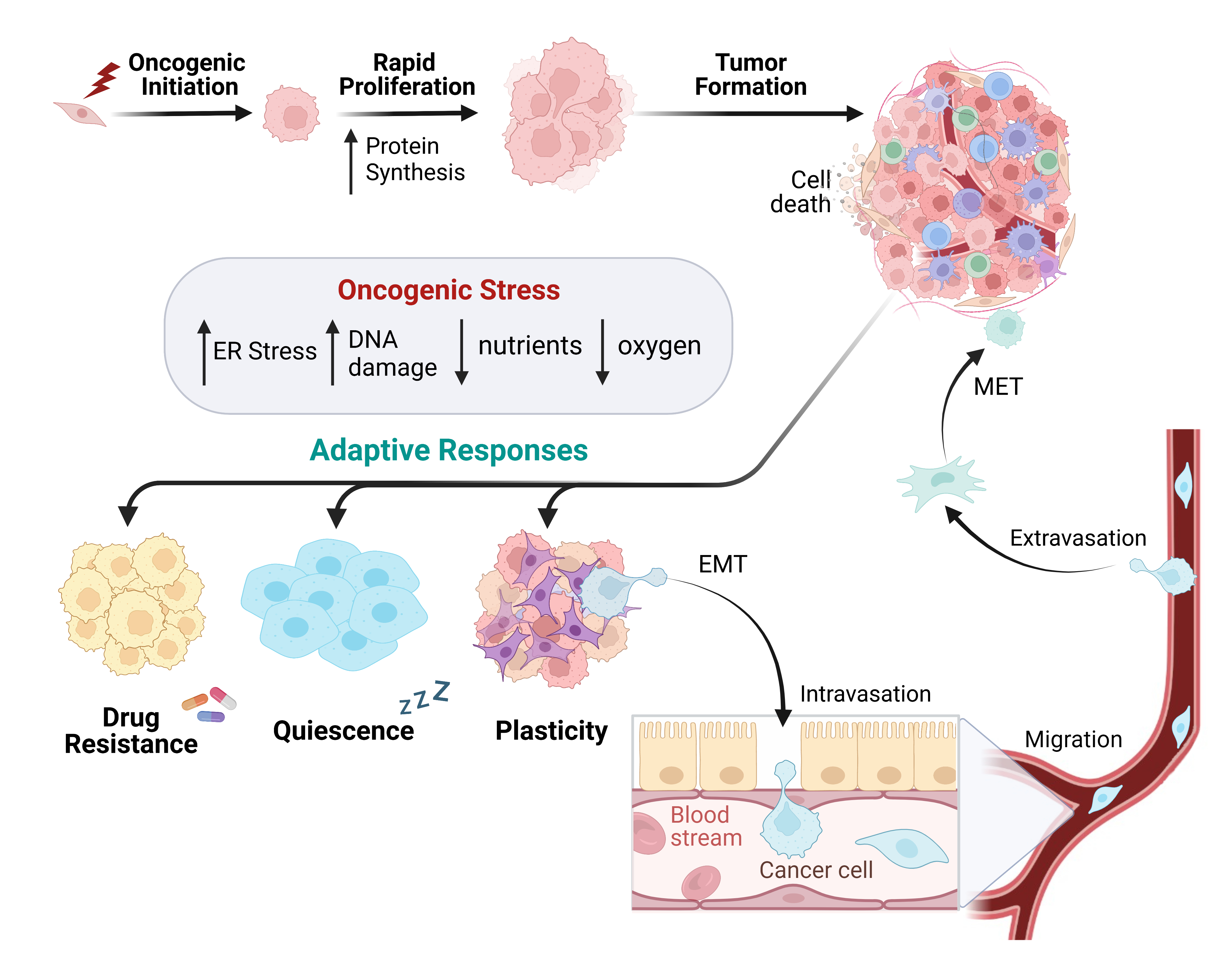
Cancer cells utilize mRNA translational control to rapidly alter gene expression for cellular advantage. In alignment with this, many key tumor suppressors and oncogenes, including KRAS and MYC, augment ribosome biogenesis and protein synthesis rates for tumor growth. Dysregulation of these genes can therefore generate aberrant networks of translational regulators, altering the proteome during cancer development. Acceleration of protein biosynthesis for growth relies heavily on adaptive processes, many of which are undetectable by genomic screening methods. These are fundamental processes for cancer cell survival caused by increased oncogenic stresses (e.g. metabolic and oxidative) during cellular transformation and progression. The Conn Lab studies the signaling cascades and regulatory mechanisms that rewire mRNA translation for survival benefit.
One of the most nutrient-depleted and hypoxic tumor types, due to low vascularity and dense stroma, is pancreatic ductal adenocarcinoma (PDAC). As a result, PDAC cells divide slowly allowing resistance to chemotherapies. This can create a worse prognosis for patients with limited treatment options. The Conn Lab is mimicking the tumor microenvironment (TME) in human cell cultures lines to study these post-transcriptional adaptations to nutrient and oxidative stress. We have identified adaptive signaling advantages used in PDAC cells for survival to TME stress and chemotherapies. We are characterizing these signaling effects and regulators to study in mouse models of PDAC. By evaluating the post-transcriptional regulation of the cancer landscape at the level of select RNA translation, we aim to improve our biological understanding of cancer progression, drug resistance, and cell state changes. Our multidisciplinary approach is designed to connect unbiased sequencing with proteomic profiles, identifying spatial RNA-protein contexts, to decipher the complete cancer proteome and the features of regulation. We aim to create efficient innovative treatments for disease progression by studying the hidden determinants of gene expression found within RNA regulation.
Constantinos Koumenis, PhD
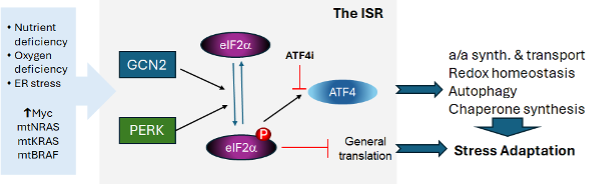
We are focused on two main areas of investigation relating to translational oncology. In the first area, we are delineating the mechanisms by which solid tumors, including pancreatic adenocarcinoma (PDAC), adapt to both cell-intrinsic stress (e.g., oncogene dysregulation) and cell-extrinsic stress (e.g., nutrient limitation and hypoxia), with an emphasis on the Integrated Stress Response (ISR). We are identifying key nodes of ISR signaling in both tumor cells and the tumor stroma and we then develop molecular approaches to target these nodes to delay tumor growth and metastasis and improve survival. The second area of interest is focused on the development of combinations of radiotherapy with engineered CAR-T cells, including the identification of novel, radiation-induced neoepitopes to improve the therapeutic response of CAR-Ts against PDAC and to translate these to the clinic.
Ellen Puré, PhD
Cancer is a manifestation of loss of tumor suppressor mechanisms and gain of oncogenic mechanisms intrinsic to the cells that undergo malignant transformation as well as extrinsic mechanisms mediated by local or systemic non-transformed cells that define the tumor microenvironment and ecosystem. Extrinsic factors associated with chronic inflammation and fibrosis can drive tumorigenesis with obesity, ageing, smoking, and chronic pancreatitis associated with increased risk of developing pancreatic cancer. Moreover, malignant cells drive the co-evolution of inflammatory, fibrotic, neural and vascular responses in tumors as well as matrix remodeling that in turn regulate malignant cell behavior. Pancreatic cancer is associated with a particularly robust desmoplastic response characterized by the expansion and activation stromal cells and dynamic remodeling of the extracellular matrix that account for a significant portion of tumor mass in pancreatic ductal adenocarcinomas (PDAs). The desmoplastic response has a net pro-tumorigenic effect due to biochemical and biomechanical signals that promote malignant cell growth, invasion and metastasis their suppress anti-tumor immunity, and confer resistance to therapy. Importantly, the microenvironment of, and the systemic response to the cancer play essential roles in the colonization and growth of metastatic tumors in distal organs, the most common cause of pancreatic cancer associated mortality. We are defining the stroma-dependent mechanisms of disease initiation, progression ad tumor resistance and developing stroma-targeted therapies to complement malignant-cell targeted therapies.
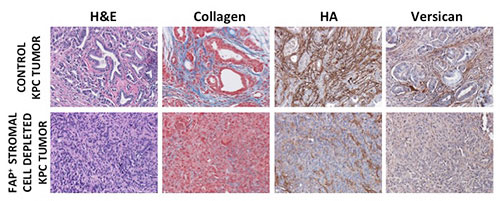
Dr. Puré and her colleagues are defining the cellular and molecular basis of the anti-tumorigenic properties of the normal healthy pancreas and the pro-tumorigenic alterations associated with ageing and obesity and the initiation and progression of pancreatic cancer. They have defined phenotypic and functional heterogeneity amongst pancreatic stromal cells that explain the potential for stroma to have opposing effects on tumor progression, and identified stromal cells required for the desmoplastic response and vascularization of PDA. They are investigating stroma-dependent pro-inflammatory mechanisms and the mechanisms by which stroma exerts its suppressive effects on anti-tumor immunity. Based on their findings, they are developing stroma-targeted approaches to complement malignant cell-targeted modalities for the treatment of PDA.
Sydney M. Shaffer, MD, PhD
The Shaffer laboratory investigates how cancer cells evolve over time, tracking the critical transitions that allow tumors to grow, spread, and resist treatment. We follow individual cancer cells and their descendants to understand key evolutionary steps such as acquisition of new mutations, gains in fitness, adaptation to therapy, and development of resistance. To trace how cancer cells are related, we leverage naturally occurring mutations as genetic barcodes in patient samples and engineered lineage tracing in cell lines and mouse models.
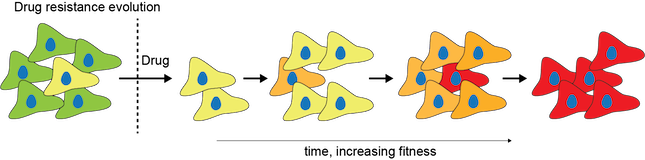
By integrating these approaches with single-cell and spatial transcriptomics, we simultaneously capture gene expression, cellular relationships, and tissue architecture. This reveals not only the molecular programs driving tumor evolution, but also how interactions with immune cells, fibroblasts, and other microenvironment components shape which cancer cell lineages survive and acquire aggressive traits as tumors progress from early lesions to metastatic disease. Our goal is to identify therapeutic vulnerabilities and predict which tumors are most likely to progress or resist treatment.
M. Celeste Simon, PhD
Dr. Simon’s laboratory studies responses to changes in oxygen (O2) availability in conjunction with nutrient scarcity and their impact on multiple diseases including certain forms of cancer. These include cancers of the kidney, liver, muscle, and pancreas. Pancreatic Cancer, a highly desmoplastic malignancy, often exhibits unusually dysfunctional blood vessels and a significant number of fibroblasts, inflammatory cells, and even neuronal cells. As such, these tumors are considered the most severally O2 limited (hypoxic) of all human cancers. Of note, hypoxia usually correlates with more aggressive cases and metastasis. The Simon laboratory is employing genetic tools to disrupt oxygen sensing pathways, including those regulated by hypoxia inducible factors (HIFs), ER stress, and metabolism to define the impact of O2/ nutrient deprivation on pancreatic cancer progression, inflammation, fibrosis, and metastasis. Current efforts are linking the relationship of O2 availability and lipid homeostasis whereby pancreatic cancer associated fibroblasts (CAFs) provide a reservoir of limiting fatty acids especially those with unsaturated fatty acyl chains. Interestingly, pancreatitis predisposes individuals to developing pancreatic ductal adenocarcinoma (PDAC) possibly because pancreatitis also results in vascular injury and hypoxia. Dr. Simon and her team are currently discerning whether hypoxia provides a mechanistic link between acute and/or chronic pancreatitis and the likelihood of pancreatic cancer initiation. Moreover, the relationship of lipid metabolism to disease progression is currently unclear and the lab hopes to provide additional mechanistic underpinnings of the “relay system” between CAFs and PDAC cells. The overall goal is to develop novel therapeutics to combat both pancreatitis and PDAC based on inhibition of a variety of O2 sensitive processes.
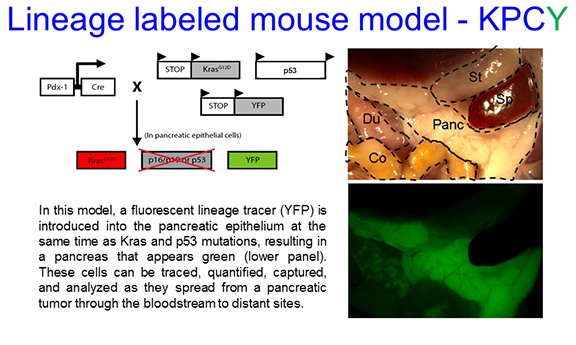
Ben Z. Stanger, MD, PhD
The Stanger laboratory relies on a mouse model of pancreatic cancer in which the animals predictably develop tumors that closely resemble human pancreatic tumors. The model – originally created at Penn and the most widely used model of pancreatic cancer progression – incorporates mutations in the Kras and p53 oncogenes, two of the “signature” mutations of pancreatic cancer. Through genetic engineering, the Stanger laboratory has introduced a gene encoding Yellow Fluorescent Protein (YFP) into the genome of the cancer prone mice, a so-called “lineage tracer” which makes the pancreatic cells turn green as the tumor is developing. Microscopes and other instruments that are capable of detecting these fluorescent cells permits them to be tracked as they acquire more malignant features and spread to distant organs (“metastasis”). Using this platform, the laboratory has made several important discoveries regarding pancreatic cancer progression and metastasis. First, pancreatic epithelial cells undergo a shift to a more motile and invasive cell type through a process known as epithelial-to-mesenchymal transition (EMT), observable with the help of the lineage tracer. Second, pancreatic cells leave the pancreas and enter the circulation at a very early time-point in tumor progression, at the PanIN stage, before cancer is detectable to a pathologist. This observation is consistent with the clinical observation that most pancreatic cancer patients already have distant metastases at the time of detection. Third, the myofibroblast stroma – a non-cancerous cellular component of a tumor – slows rather than accelerates tumor growth, as its elimination led to faster growing tumors. Again, this finding is consistent with the results of clinical trials using hedgehog inhibitors, which deplete the myofibroblast stroma but unfortunately led to a worse clinical outcome.
The lab’s ongoing studies concern molecular mechanisms of invasion and metastasis. In one project, we are identifying the mechanisms of EMT in this model, providing the first molecular insights into this cellular shift in the context of a naturally progressing tumor. Another project is concerned with the cellular and molecular determinants of metastasis. We have found that the tumors which arise in the mouse model vary with respect to their ability to spread, and we hope to identify the genes that enable spread so that they may be targeted. Another collaborative project seeks to identify material circulating in the blood (cells, DNA or small vesicles) that could be used as a diagnostic test for incipient pancreatic cancer. This project employs mouse samples but insights will be rapidly translated to human specimens. Finally, we are engaged in a number of preclinical trials in collaboration with academic and industry partners to test promising compounds for the treatment of pancreatic cancer that have clinical potential.
Ioannis I. Verginadis, MSc, PhD
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest cancers, with a 5-year survival rate of about 13%, mainly due to late detection, rapid metastasis, and resistance to therapy. The dense and desmoplastic tumor microenvironment (TME) of PDAC plays a significant role in its development and metastasis. The Verginadis lab focuses on identifying gene signatures, signaling pathways, and cell populations that contribute to this complex and treatment-resistant TME. Our multi-step approach includes: 1) Using cell lines derived from KPC-YFP tumor models (from Stanger’s lab) to perform functional assays in response to chemo/radiation; 2) Employing preclinical PDAC models to characterize the TME in response to chemo/radiation; 3) Integrating AI-driven analysis of our data and publicly available scRNA-sequencing and spatial transcriptomics from mouse and human studies. This step aims to define and predict patterns of treatment resistance in cancer-associated fibroblasts (CAFs), their dynamics within the TME, and interactions with other TME cell populations; 4) Using a 3D co-culture system of patient-derived organoids (PDO) (from Johns Hopkins Hospital) with patient-derived CAFs to study their dynamics and responses to chemo/radiation; 5) Conducting a retrospective analysis of clinical data alongside transcriptomics (bulk RNA-seq) and whole exome sequencing of resected PDAC tumors to identify gene signatures that predict response to treatment (chemo/radiation) and metastasis potential.

Robert H. Vonderheide, MD, DPhil
Pancreatic cancer is well known for its very poor prognosis, a situation that this Center is dedicated to changing. The disease is highly refractory to standard therapy, especially for patients who present with metastatic disease. Unfortunately, the “statistics” are getting worse. Pancreatic cancer is one of only two cancers among 21 histotypes for which the death rate in the United States rose from 1990 to 2008 (the other is liver/intrahepatic bile duct cancer). Pancreatic cancer is likely to become the second leading cause of cancer-related death in the United States by 2020 (behind lung cancer, which is declining). Although new combination chemotherapies are able to stabilize many patients who present with metastatic pancreatic cancer (representing a major clinical advance), response rates remain <35%, and patients with long-term complete remissions after treatment with these therapies are rare.
In the context of this unmet clinical need, genetically engineered murine models of pancreatic neoplasia have become an increasingly important tool for us to use to garner biological insights that might lead to novel therapeutic strategies. Based largely on the selective expression of oncogenic Kras in the pancreas of immune-competent hosts, these models reproduce key biological aspects of the human disease, including its highly desmoplastic and inflammatory tumor microenvironment. These models offer the opportunity to uncover novel “non-tumor cell-autonomous” therapeutic targets in the tumor stroma, which can be tested for efficacy in tumor-bearing mice prior to translation to the clinic. Because pathology in these models advances from pre-invasive disease to invasive disease, agents that prevent or reverse the earliest neoplastic events can also be explored.
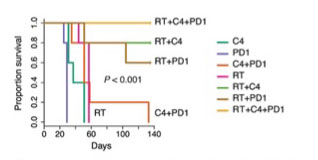
Survival curves following treatment with various forms of immunotherapy
The Vonderheide Laboratory is particularly focused on finding new immune therapies for pancreatic cancer. In this form of cancer, a massive infiltration of immunosuppressive leukocytes into the tumor stroma is an early and consistent event in oncogenesis. In our mouse models, intratumoral effector T cells are rare, as they are in the majority of patients with pancreatic cancer. This pathophysiology is in contrast to many other solid tumors for which infiltration of effector T cells is often prominent, associated with improved clinical outcomes, and mechanistically contributes to tumor immunoediting that ultimately can mediate immune escape. In pancreatic cancer, increasing evidence suggests that an inflammatory program establishes immune privilege in the tumor microenvironment. Indeed, pancreatic tumor cells might remain intrinsically sensitive to T cell killing because they have never been exposed to Darwinian-like T-cell-selective pressure in vivo. In support of this hypothesis, recent studies demonstrate that derailing immune suppressive pathways in the pancreatic cancer microenvironment, such as tumor-derived GM-CSF, facilitates T-cell-mediated tumor rejection. In addition, various combinations of immune therapy and standard therapy can cooperate to kill pancreatic cancer cells, including blockade of checkpoint molecules such as CTLA-4 and PD-1, activation via CD40, and standard therapies such as radiation and chemotherapy. Moreover, because mutant Kras appears capable of orchestrating a tumor-promoting microenvironment beyond well-described tumor-cell-autonomous Kras mechanisms, pharmacological inhibition of oncogenic Kras and pathways downstream, therefore, might realistically be expected to derail these tumor-promoting non-cell-autonomous mechanisms, providing even more incentive (if more were needed) for renewed efforts to develop drugs for Kras.
Kathryn E. Wellen, Ph.D.
Metabolism is extensively reprogrammed in cancer cells to support growth and proliferation and to allow cells to adapt to nutrient and oxygen deprived microenvironments, which are characteristic of pancreatic tumors. Worryingly, the rate of new pancreatic cancer cases has been rising in recent decades. Obesity is associated with an increased risk of developing pancreatic cancer, highlighting the importance of understanding the influence of systemic metabolic perturbations on pancreatic tumors. The Wellen lab is investigating how systemic metabolic factors impact pancreatic cancer development and progression. We are currently using mouse models to study the interplay between adipose (fat) tissue and pancreatic tumors. We are also investigating the roles of specific nutrients, such as branched chain amino acids, in tumor development and growth. We aim to decipher mechanisms that contribute to tumor formation, towards the goal of reducing pancreatic cancer incidence. We also aim to better understand how nutrient metabolism in cancer cells and in non-malignant cells in the microenvironment impacts tumor growth and progression, towards the goal of identifying improved therapeutic strategies.
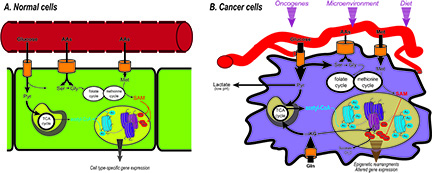
A) Metabolites such as acetyl-CoA and S-adenosylmethionine (SAM) are substrates of chromatin modifying enzymes. B) In cancer cells, metabolic reprogramming can alter the availability of these metabolites and contribute to cancer cell epigenetic alterations. (Figure from: Carrer and Wellen, Curr Opin Biotech, 2015)
Kenneth S. Zaret, PhD
Pancreatic ductal adenocarcinoma (PDAC) is usually detected too late for efficient intervention to stop the disease. Our laboratory, in conjunction with the liquid biomarker laboratory headed by Dr. Erica Carpenter and an IRB with Dr. Bryson Katona, is dedicated towards identifying blood biomarkers of early stage disease. If PDAC is detected early, it is possible to resect the tumor with minimal consequential metastasis. Because antibodies remain the most sensitive way to detect new marker expression, requiring only microliters of blood, we are focusing on blood protein biomarkers that can be assessed by routine screening, particularly of high-risk individuals. To date, we have used different modalities for discovering proteins that are selectively secreted or released from early stage PDAC cells, including using a human stem cell-based model that recapitulates early PDAC development (PMC3726210). More recently, we have obtained different carefully curated sets of blood samples from individuals with early stage PDAC and have used mass spectrometry to compare the expressed proteins with proteins seen in various control samples and samples of late stage PDAC. While the field to date has focused biomarker discovery on late stage PDAC samples that can be most conveniently obtained, our hope is that by directly studying early stage samples, we can discover biomarkers that will perform the best in early stage detection.
Rong Zhou, PhD
Rong Zhou, PhD, leads a translational molecular imaging laboratory focused on developing quantitative PET and MRI biomarkers that can be integrated with novel therapeutic interventions targeting reprogrammed metabolism and other oncogenic pathways in cancer, including pancreatic ductal adenocarcinoma (PDAC). Her team led the preclinical development of glutamine PET using [18F]-(2S,4R)-4-fluoroglutamine (F-Gln PET) to interrogate glutaminolysis, a metabolic program frequently rewired in aggressive tumors and linked to glutamine dependence. The Zhou lab applies a rigorous bench-to-bedside pipeline—from molecular and cellular validation to animal models, IND-enabling studies, and clinical translation. In PDAC models, her group demonstrated that clinically deployable dynamic contrast–enhanced (DCE) MRI detects early responses to stroma-directed therapy through changes in perfusion and permeability. More recently, the lab developed multiparametric imaging markers that provide early evidence of KRAS inhibitor target engagement and capture acquired resistance during prolonged treatment. Dr. Zhou is a PI of the Penn Pancreatic Cancer Imaging Resource within the NCI’s Co-Clinical Imaging Research Resources Program (https://dctd.cancer.gov/research/networks/cirp), harmonizing imaging endpoints across matched preclinical and clinical studies through close collaboration with clinicians and industry partners.
