Our Research
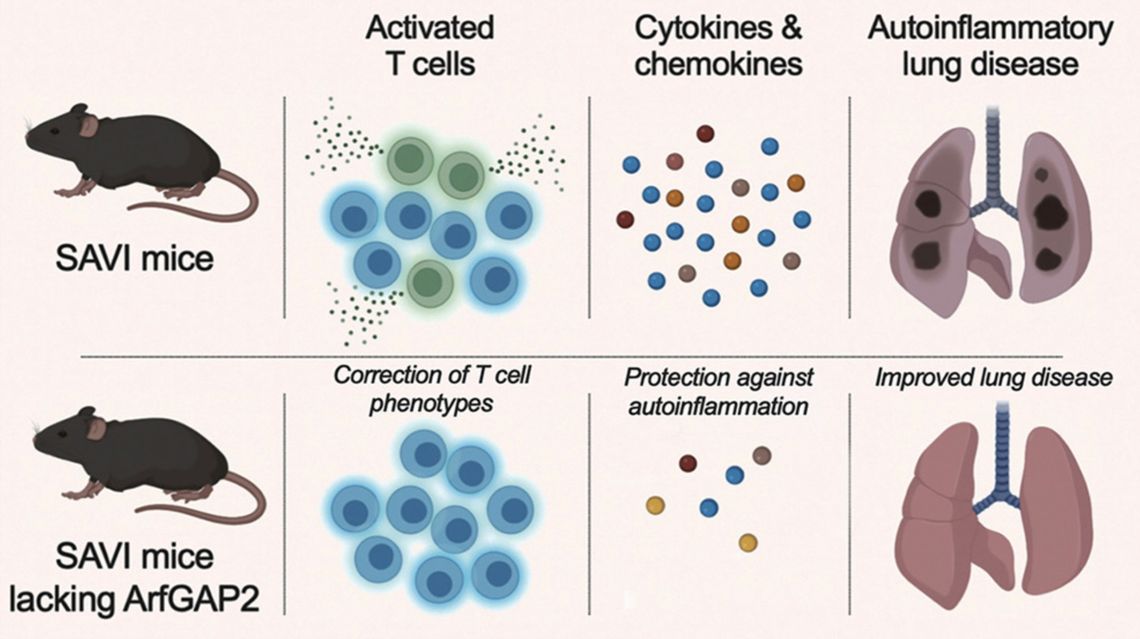
Read some of our latest work on STING and a rare disease known as SAVI, recently published in Cell
Read some of our latest work on TREX1 and a rare disease known as RVCL, recently published in Nature Communications
Why we study rare diseases
A major priority for our laboratory is to define mechanisms of rare rheumatic diseases, and to develop and test personalized therapies for those diseases. In particular, we focus our studies on cytosolic nucleic acid-sensing pathways. We currently have active collaborations with multiple investigators to develop personalized medicines for rare diseases. This includes collaborators at multiple institutions around the country.
Rare disease research is important because these diseases are devastating and untreatable. Many of these patients die prematurely, but their diseases could be cured since they are a consequence of a single gene mutation.
Studying monogenic diseases also gives insight into fundamental mechanisms of biology and disease. Another benefit of this work is that the pathways targeted are highly relevant for a wide variety of common diseases, including infections, cancer, and autoimmune diseases. Thus, many of the therapies being developed are likely to be of broad utility for common diseases as well.
Read more about specific projects by clicking the menus below.
Rare disease discovery and mechanisms
Our laboratory is currently supported by the National Institutes of Health (NIH), by private foundations, and by rare disease patients, as well as by their friends and families.
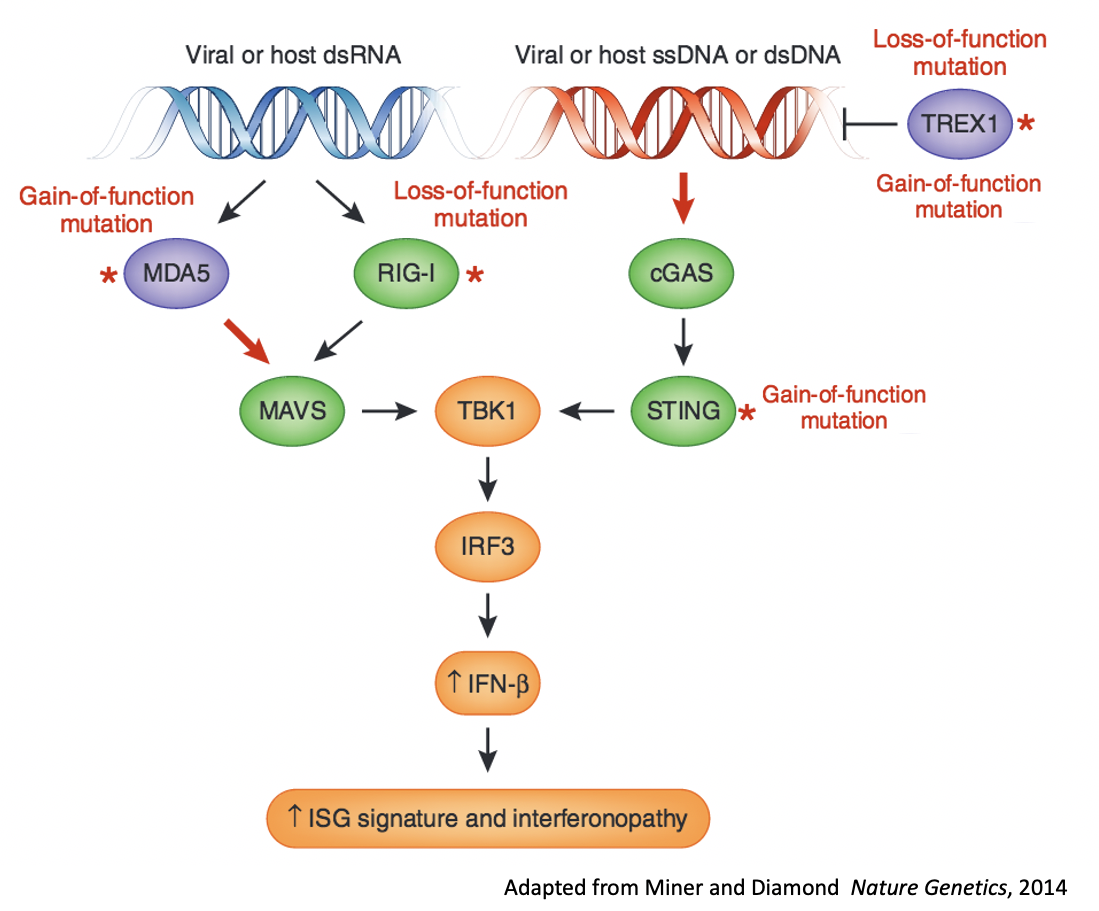
Our research is centered on immunological pathways related to nucleic acid sensors that reside in the cytosol. These pathways play a vital role as he first line of defense against viral infections. Activation of nucleic acid sensors leads to induction of interferons. Mutations in genes that encode nucleic acid sensors can cause autoimmune disease, including a class of diseases known as "interferonopathies."
Our laboratory merges combined expertise in immunology, rheumatology, and virology to elucidate mechanisms of rare diseases, and to understand how nucleic acid sensors regulate immunity against viruses. Additionally, we are working with collaborators to describe novel diseases related to mutatiions in these pathways.
We also study how these pathways regulate the response to viral infections, and we work with many different types of viruses in the laboratory, including with Biosafety Level 3 (BSL3) viruses.
Role of TREX1 in RVCL and vasoocclusion
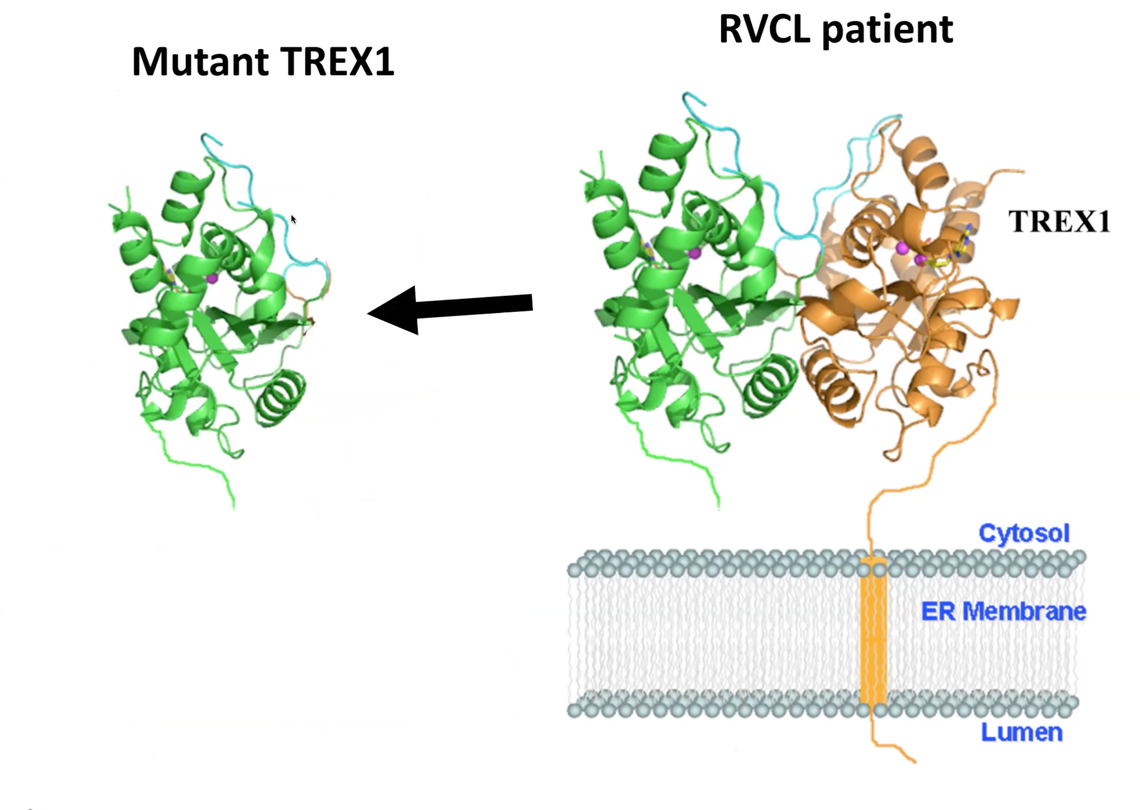
RVCL is a rare inherited disease caused by mutations in a gene called TREX1, which is found on chromosome 3. TREX1 is a DNAse that negatively regulates the cGAS-STING pathway, which is one of the cytosolic nucleic acid-sensing pathways that we study in our lab.
RVCL is inherited in a autosomal dominant pattern, which means that about half of family members are affected. Patients with RVCL have one normal copy of TREX1, and one mutated copy of TREX1.
Normally, the TREX1 protein is attached to a membrane inside the cell. In patients with RVCL, one copy of TREX1 is abnormally shortened because of the mutation. This causes the TREX1 protein to float freely in the cell, rather than being anchored to the membrane.
Mislocalization of the TREX1 protein causes cellular damage and disease. We recently discovered the mechanism underlying the disease. Additionally, we have developed novel therapies targeting the mutated TREX1 gene and protein, with the goal of starting clinical trials at Penn in the near future.
L earn more about RVCL by exploring these links:
earn more about RVCL by exploring these links:
Interferon (IFN)-mediated immunity against viruses
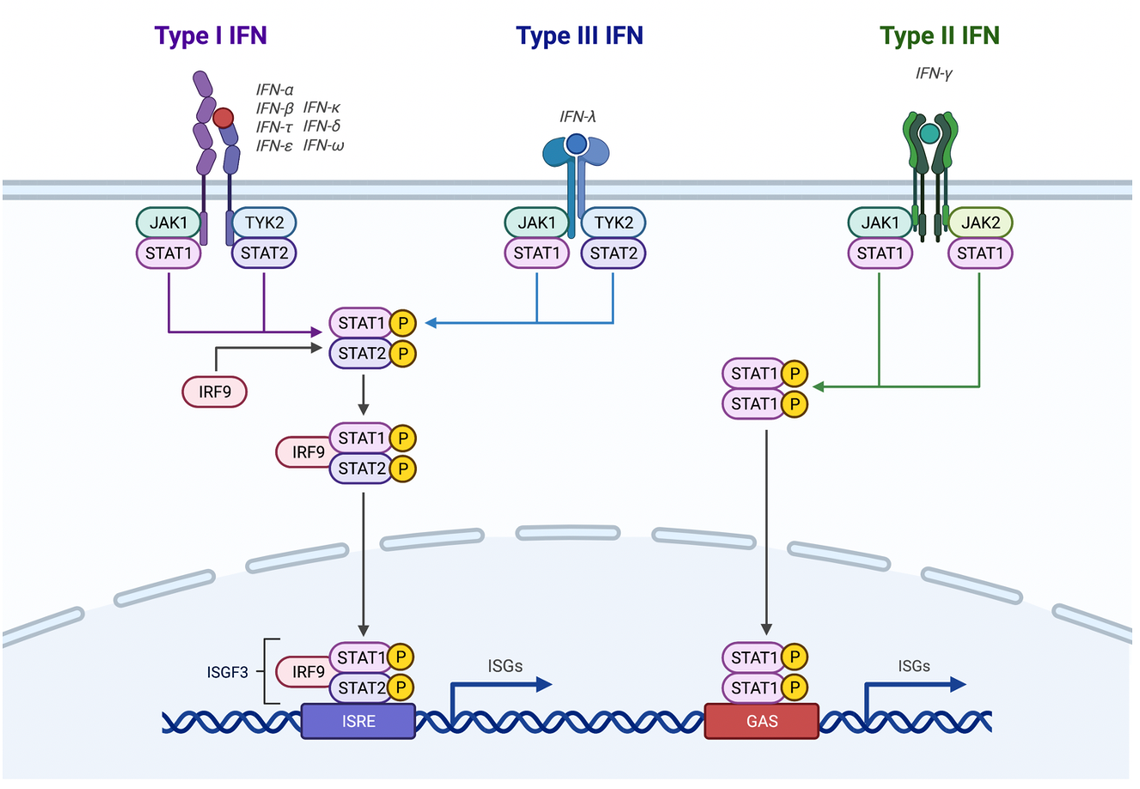
We have conducted multiple NIH-funded projects related to interferon (IFN)-mediated immunity against viruses. Several trainees in our laboratory have done work in this area.
This led to multiple publications in Science Translational Medicine, Cell Reports, and Journal of Virology.
Some of our recent discoveries include the identification of the type III interferon receptor (IFN-lambda receptor) in the human cornea, and the discovery that multiple flaviviruses including West Nile virus and Powassan virus can cause congenital infection in model systems. Additionally, we found that the human cornea does not support growth of SARS-CoV-2 in culture conditions, although that finding does not necessarily indicate that the eye or surrounding tissues would not be a site of initial infection by this virus.
CRISPR/Cas9 screening to identify novel regulators of innate immune pathways
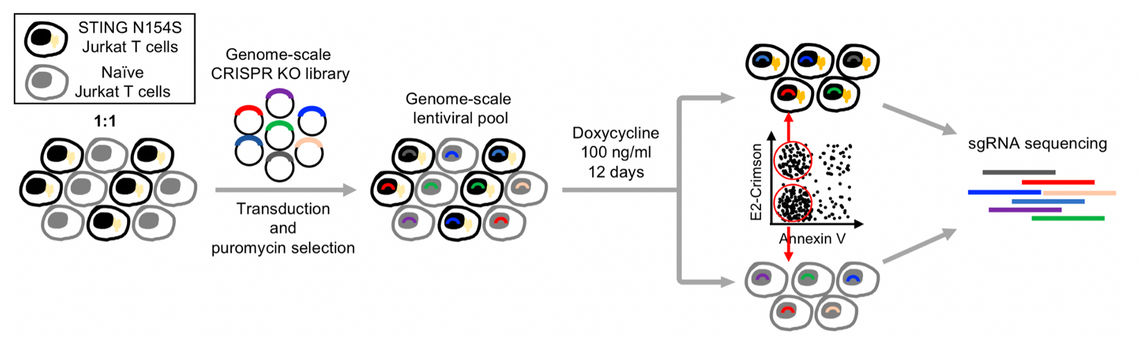
A major focus of the work in our laboratory has been to use patients' disease-causing mutations to identify novel regulators of nucleic acid-sensing pathways. This work has major implications for identifying therapeutic targets. Additionally, this approach has led us to discover disease-specific regulators of signaling. Studying patient mutations also gives insight into normal biology because it helps to clarify mechanisms by which normal genes and proteins are regulated.
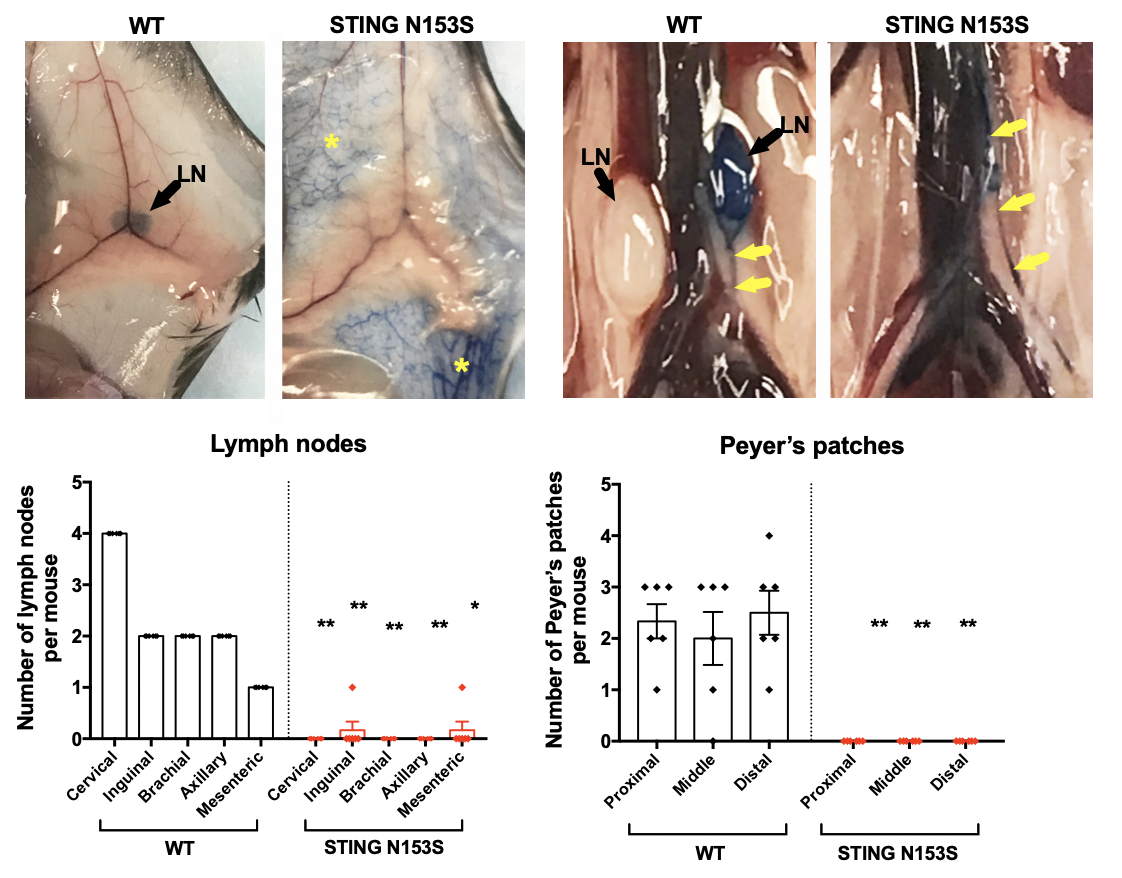
Interferon-mediated regulation of lymph node development