Our Research
Our lab has a long-term interest in understanding the fundamental aspects of skeletal muscle and cardiac function in normal or diseased conditions and in the practical aspects of manipulating these functions by using animal models and tissue engineering approaches for treatment intervention.
We believe that combining bioengineering approaches with stem cell biology, testing cell-based therapies and modulating the signaling pathways of adult stem cells we can enhance the replacement of skeletal muscle and cardiac tissue upon injury. We are presently combining genetic knockout mice and gene recombination with cellular engineering techniques to investigate the temporal and spatial communication of cells and signaling pathways during muscle regeneration.
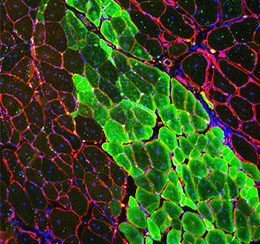
Disruption of skeletal muscle integrity results in muscle atrophy and degeneration but the cellular and molecular underpinnings that will promote regeneration remain elusive. We showed that inhibition of NF-κB pathway induces regeneration after injury and protects against denervation-induced muscle atrophy (JCI, 2016). Furthermore, our research revealed a muscle stem cell-autonomous increase in the activity of the p38 mitogen-activated protein kinase pathway (Nature Medicine, 2014) and provided the first evidence that polarization of macrophages (from pro-inflammatory to anti-inflammatory) is fundamental for skeletal muscle regeneration in mice (PNAs, 2009). Previously, we developed a new mouse model (dystrophic mice with shorten telomeres, mdx/mTR mouse) that better replicates the phenotype of Duchenne Muscular Dystrophy (DMD), providing novel evidence that the skeletal muscle phenotype is caused by a failure of skeletal muscle stem cells to maintain the damage-repair cycle initiated by dystrophin deficiency (Cell, 2010).
Current Research
Our current research has 3 connected thematic areas:
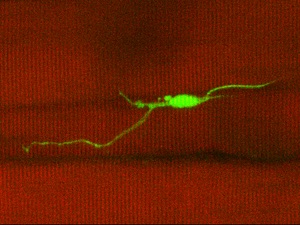
Mechanobiology and morphological heterogeneity in muscle stem cells. A first research focus is understanding the role of stem cell behavior within their endogenous tissue environment in steady conditions and how their performance changes under injury and/or diseased conditions. For this purpose, we developed a new mouse model (Pax7EGFP), where EGFP expression is both robust and dynamic and a real-time indicator of MuSC stemness (Skeletal Muscle, 2018). Utilizing this genetic tool in combination with our new in vivo 2-photon microscopy technique to monitor stem cells for the first time in real-time, we uncovered a surprising finding: MuSCs in vivo are not round cells, as previously believed, but rather exhibit morphological heterogeneity, displaying a variability in the number and shape of axon-like cellular extensions, which we have termed protrusions (Star Protocols, 2023). In contrast to the earlier assumptions that quiescence is a passive and dormant state devoid of activities, our work reveals that cellular quiescence is actively maintained and that it corresponds to a collection of morphological heterogeneous stem cell states that we classified as responsive (“primed” to respond to an injury), intermediate, or sensory cells (more “dormant” genuine state). While we found that stem cell protrusions do not mobilize cells under steady-state conditions, they play an important role in sensing both the stem cell niche and the surrounding tissue (Science Advances 2022). Our lab has taken new approaches to understand how resident stem cells convert mechanical forces into biological signals to provide cells with information about their microenvironment. We further identified a mechanosensing pathway as a key regulator for modulating stem cell states during repair, and its pharmacological reactivation ameliorates disease morphology in Duchenne Muscular Dystrophy (DMD) muscles. Altogether, these pioneering studies have changed the classical conception of dormancy of adult resident stem cells and provided a paradigm for quiescence and activation during physiological and pathological conditions. Such studies provide us with the unique ability to address fundamental questions into the emerging cell biology of stem cells.
Telomere biology in cardiac and skeletal muscles. The second research theme is on Duchenne Muscular Dystrophy (DMD), the most common inherited form of myopathy, affecting both skeletal and cardiac muscles. Repeated cycles of muscle damage and repair in DMD lead to stem cell dysfunction in skeletal muscles. Using a powerful and innovative single-cell imaging technique we developed to monitor diseased adult muscle stem cells (MuSCs), we discovered that telomere shortening is a key feature of dystrophic MuSCs in both mice and DMD patients, starting at a very young age (Stem Cell Reports, 2018). Importantly, we discovered the underlying mechanism behind such molecular alterations. We demonstrated for the first time that prolonged NF-κB activation in diseased mouse MuSCs leads to shortened telomeres, independent of cell replication, via a mechanism that involves Ku80 alterations and excessive DNA damage to telomeres. As a result, there is premature telomere uncapping in diseased MuSCs leading to stem cell exhaustion (Cell Reports, 2021). Since the major cause of death in DMD is cardiac failure, we extended our studies to heart tissue and measured telomere length in a cell-type specific manner that bypasses previous limitations by avoiding contributions of undesired cell types in cardiac tissues (Nature Protocols, 2017). We discovered an important mechanistic role for telomeres in human cardiomyopathy and demonstrated shortened telomeres only in cardiomyocytes of DMD patients. Furthermore, we provided the first evidence of a direct association between cardiomyocyte-specific telomere shortening and human heart failure, highlighting its significance in aging and gender and demonstrated that telomere shortening is a previously unrecognized signature of cardiac diseases (J Am Heart Assoc, 2017).

and heterotopic bone formation
Communication of muscle progenitor cells (MuSCs-FAPs) during regeneration. In a third and related research project, we investigated the dynamics of two adult resident stem cells (MuSCs and Fibro-adipogenic progenitors, also called FAPs) during skeletal muscle regeneration. We identified for the first time that Gli1 (a crucial mediator of Hh signaling), labels a distinct subset of FAPs. We discovered that in the absence of Gli+FAPs, skeletal muscle repairs poorly with abnormal intermuscular accumulation of fat (JBMR, 2021). We have extended these studies to address a major challenge of muscle regeneration in a rare disease called Fibrodysplasia ossificans progressiva (FOP), in which upon injury, heterotopic (extra-skeletal) bone forms within skeletal muscle. Initially, we showed that elevated BMP signaling alters cellular mechanotransduction and muscle microenvironment after injury (J Bone Miner Res, 2019). More recently, we demonstrated that FOP MuSCs fail to proceed normally during myogenesis, due to an improper communication with FAPs during muscle regeneration. These data reveal a fundamental role of FAPs in influencing the myogenic activity of MuSCs in this rare disease after injury and, as a consequence, MuSC-FAP coordination may be an important target for future therapeutic interventions to improve the health of those with FOP (NPJ Regen Med, 2021, mdpi Biomedicines, 2024, PNAs, 2025). Importantly, we found that FAPs are the predominant cell-of-origin for HO formation, recapitulating the human pathology. This is an ideal system to identify intrinsic properties and signals affecting cell-cell communication among different resident stem cells as well as is a model for pre-clinical studies.